мышечная дистрофия Дюшенна
| мышечная дистрофия Дюшенна | |
|---|---|
 | |
| Микроскопическое изображение поперечного сечения икроножной мышцы человека с мышечной дистрофией Дюшенна, показывающее обширное замещение мышечных волокон клетками жировыми . | |
| Произношение | |
| Специальность | Детская неврология , нервно-мышечная медицина , медицинская генетика |
| Симптомы | Мышечная слабость , проблемы со стоянием, сколиоз. [2] [3] [4] |
| Обычное начало | Около 4 лет [2] |
| Причины | Генетический ( Х-сцепленный рецессивный ) [3] |
| Метод диагностики | Генетическое тестирование [3] |
| Уход | Фармакологическое лечение, физиотерапия , брекеты , логопедия, эрготерапия, хирургия, вспомогательная вентиляция [2] [3] |
| Медикамент | Кортикостероиды |
| Прогноз | продолжительность жизни : 28–30 лет |
| Частота | У мужчин – 1 из 3500–6000. [3] У женщин – 1 на 50 000 000. [5] |
Мышечная дистрофия Дюшенна ( МДД ) — тяжелый тип мышечной дистрофии, поражающий преимущественно мальчиков. [3] [6] [7] Мышечная слабость обычно начинается примерно в возрасте четырех лет и быстро прогрессирует. [2] Первоначально потеря мышечной массы происходит в бедрах и тазе , распространяясь на руки. [3] что может привести к трудностям при вставании. [3] К 12 годам большинство людей с мышечной дистрофией Дюшенна не могут ходить. [2] Пораженные мышцы могут казаться больше из-за увеличения содержания жира . [3] и сколиоз является распространенным явлением. [3] Некоторые люди могут испытывать умственную отсталость , [3] а у женщин, несущих единственную копию мутировавшего гена, могут проявляться легкие симптомы. [3]
Мышечная дистрофия Дюшенна вызвана мутациями и/или делециями в любом из 79 экзонов, кодирующих большой дистрофин белок , который необходим для поддержания целостности клеточной мембраны мышечного волокна . [3] Заболевание следует по Х-сцепленному рецессивному типу наследования: примерно две трети случаев наследуются от матери, а одна треть возникает в результате новой мутации . [3] Диагноз часто можно поставить при рождении с помощью генетического тестирования , а повышенный уровень креатинкиназы в крови указывает на заболевание. [3]
Несмотря на отсутствие известного лечения, такие стратегии лечения, как физиотерапия , брекеты и корректирующая хирургия, могут облегчить симптомы. [2] Вспомогательная вентиляция может потребоваться пациентам со слабостью дыхательных мышц . [3] В настоящее время доступно несколько препаратов, предназначенных для устранения основной причины, включая генную терапию ( Элевидис ) и антисмысловые препараты ( Аталурен , Этеплирсен и т. д.). [3] Другие используемые лекарства включают глюкокортикоиды ( Дефлазакорт , Ваморолон ); блокаторы кальциевых каналов ( Дилтиазем ); для замедления дегенерации скелетных и сердечных мышц, противосудорожные средства для контроля судорог и некоторой мышечной активности, а также ингибиторы гистондеацетилазы ( гивиностат ) для задержки повреждения умирающих мышечных клеток . [2] [3]
Приводятся различные цифры встречаемости мышечной дистрофии Дюшенна. Один источник сообщает, что при рождении он поражает примерно одного из 3500–6000 мужчин. [3] Другой источник сообщает, что мышечная дистрофия Дюшенна является редким заболеванием и встречается с частотой 7,1 на 100 000 новорожденных мальчиков. [8] Ряд источников, упомянутых в этой статье, указывают на частоту 6 случаев на 100 000 человек. [9]
Мышечная дистрофия Дюшенна – наиболее распространенный тип мышечной дистрофии. [3] со средней продолжительностью жизни 28–30 лет. [10] [11] Однако при комплексном уходе некоторые люди могут дожить до 30 или 40 лет. [3] Мышечная дистрофия Дюшенна у женщин встречается значительно реже: примерно у одной из 50 000 000 живорожденных девочек. [5]
Признаки и симптомы
[ редактировать ]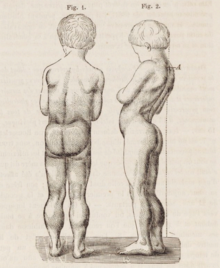
Мышечная дистрофия Дюшенна вызывает прогрессирующую мышечную слабость из-за мышечных волокон соединительной тканью или жиром. беспорядка, гибели и замены [3] , произвольные мышцы В первую очередь поражаются особенно бедер , области таза , бедер , икр . [3] [2] [12] В конечном итоге он распространяется на плечи и шею, а затем на руки , дыхательные мышцы и другие области. [12] Усталость является обычным явлением. [13]
Признаки обычно появляются в возрасте до пяти лет и могут даже наблюдаться с того момента, как мальчик делает свои первые шаги. [14] Наблюдаются общие трудности с двигательными навыками , что может привести к неловкости при ходьбе, шагании или беге. [15] Они склонны ходить на цыпочках , [15] отчасти из-за укорочения ахиллова сухожилия, [16] и потому, что он компенсирует слабость разгибателей колена. [12] Падения могут быть частыми. [17] Мальчику становится все труднее ходить. Его способность ходить обычно полностью утрачивается до 13 лет. [15] Большинство мужчин, страдающих мышечной дистрофией Дюшенна, к 21 году становятся практически «парализованными от шеи и ниже». [14] Кардиомиопатия , особенно дилатационная кардиомиопатия , встречается часто и наблюдается у половины 18-летних. [15] Развитие застойной сердечной недостаточности или аритмии (нерегулярного сердцебиения) происходит лишь изредка. [12] На поздних стадиях заболевания могут возникнуть нарушения дыхания и глотания, что может привести к пневмонии . [18]
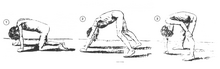
Классическим признаком мышечной дистрофии Дюшенна являются трудности с вставанием из положения лежа или сидя. [17] что проявляется положительным признаком Гауэрса . Когда ребенок пытается встать из положения лежа на животе, он компенсирует слабость мышц таза за счет использования верхних конечностей: [15] сначала вставая на руки и колени, а затем «поднимая» руки вверх по ногам, чтобы встать прямо. Другим характерным признаком мышечной дистрофии Дюшенна является псевдогипертрофия (увеличение) мышц языка, икр, ягодиц и плеч (около 4–5 лет). Мышечная ткань со временем заменяется жировой и соединительной тканью, отсюда и термин псевдогипертрофия. Могут возникнуть деформации мышечных волокон и мышечные контрактуры ахиллова сухожилия и подколенных сухожилий, которые нарушают функциональность, поскольку мышечные волокна укорачиваются и фиброзируются в соединительной ткани . [12] Могут возникнуть деформации скелета, такие как гиперлордоз поясничного отдела , сколиоз вперед , наклон таза и деформации грудной клетки. Считается, что поясничный гиперлордоз является компенсаторным механизмом в ответ на слабость ягодичных мышц и четырехглавых мышц, все из которых вызывают изменение осанки и походки (например, ограничение разгибания бедра). [19] [20]
Встречаются некостно-мышечные проявления мышечной дистрофии Дюшенна. Существует более высокий риск нейроповеденческих расстройств (например, СДВГ ), нарушений обучения ( дислексии ) и непрогрессирующей слабости определенных когнитивных навыков (в частности, кратковременной вербальной памяти). [15] которые, как полагают, являются результатом нехватки дистрофина в мозге. [21]
Причина
[ редактировать ]
Мышечная дистрофия Дюшенна обусловлена мутацией гена дистрофина, расположенного на коротком плече Х-хромосомы ( локус Xp21). [22] который кодирует белок дистрофин. Мутации могут передаваться по наследству или возникать спонтанно при передаче по зародышевой линии. [ нужна ссылка ] вызывая значительное снижение или отсутствие дистрофина, белка, который обеспечивает структурную целостность мышечных клеток. [23] Дистрофин отвечает за соединение актинового цитоскелета каждого мышечного волокна с подлежащей базальной пластинкой ( внеклеточным матриксом ) через белковый комплекс , содержащий множество субъединиц. Отсутствие дистрофина позволяет избытку кальция проникать через сарколемму (клеточную мембрану). [24]

Мышечная дистрофия Дюшенна у женщин встречается крайне редко (около 1 на 50 000 000 родившихся девочек). [5] Это может произойти у женщин с больным отцом и матерью-носителем, у тех, у кого отсутствует Х-хромосома или у тех, у кого инактивирована Х-хромосома (наиболее распространенная из редких причин). [25] Дочь матери-носителя и больного отца будет поражена или носительница с одинаковой вероятностью, поскольку она всегда унаследует пораженную Х-хромосому от своего отца и имеет 50% шанс унаследовать пораженную Х-хромосому от своей матери. . [26]
Было замечено, что нарушение гематоэнцефалического барьера является заметной особенностью развития мышечной дистрофии Дюшенна. [27]
Диагностика
[ редактировать ]Мышечную дистрофию Дюшенна можно обнаружить с точностью около 95% с помощью генетических исследований, проводимых во время беременности. [18]
ДНК-тест
[ редактировать ]Специфичная для мышц изоформа гена дистрофина состоит из 79 экзонов , а тестирование ДНК ( анализ крови ) и анализ обычно позволяют определить конкретный тип мутации экзона или экзонов, которые затронуты. ДНК-тестирование подтверждает диагноз в большинстве случаев. [28]
Биопсия мышц
[ редактировать ]Если тестирование ДНК не выявило мутацию, может быть проведена биопсия мышц. [29] Небольшой образец мышечной ткани извлекается с помощью иглы для биопсии. Ключевыми тестами, выполняемыми с образцом биопсии при мышечной дистрофии Дюшенна, являются иммуногистохимия , иммуноцитохимия и иммуноблоттинг на дистрофин, и их интерпретацию должен проводить опытный нервно-мышечный патолог. [30] Эти тесты предоставляют информацию о наличии или отсутствии белка. Отсутствие белка является положительным тестом на мышечную дистрофию Дюшенна. При наличии дистрофина тесты показывают количество и молекулярный размер дистрофина, помогая отличить мышечную дистрофию Дюшенна от более легких фенотипов дистрофинопатии . [31] За последние несколько лет были разработаны тесты ДНК, которые выявляют большее количество мутаций, вызывающих это заболевание, и биопсия мышц не требуется так часто, чтобы подтвердить наличие мышечной дистрофии Дюшенна. [32]
Пренатальные тесты
[ редактировать ]Пренатальный тест можно рассмотреть, если мать является известным или подозреваемым носителем. [33]
Перед инвазивным тестированием важно определить пол плода; в то время как мужчины иногда страдают этим Х-сцепленным заболеванием, мышечная дистрофия Дюшенна у женщин встречается крайне редко. Этого можно достичь с помощью ультразвукового сканирования на сроке 16 недель или позже с помощью тестирования свободной ДНК плода (cffDNA). Биопсия ворсин хориона (CVS) может быть сделана на сроке 11–14 недель, при этом риск выкидыша составляет 1%. Амниоцентез можно провести через 15 недель, при этом риск выкидыша составляет 0,5%. Неинвазивное пренатальное тестирование можно провести примерно через 10–12 недель. [34] Другим вариантом в случае неясных результатов генетического теста является биопсия мышц плода. [ нужна ссылка ]
Уход
[ редактировать ]
Лечения мышечной дистрофии Дюшенна не существует. [35]
Лечение обычно направлено на контроль симптомов, чтобы максимизировать качество жизни, которое можно измерить с помощью специальных опросников. [36] и включить:
- Кортикостероиды, такие как преднизолон , дефлазакорт и ваморолон (Агамри), приводят к кратковременному улучшению мышечной силы и функции на срок до 2 лет. [37] Сообщалось также, что кортикостероиды помогают продлить ходьбу, хотя доказательства этого не являются убедительными. [38]
- Физиотерапия, специфичная для конкретного заболевания, полезна для поддержания мышечной силы, гибкости и функционирования. Его цель: [39]
- Сведите к минимуму развитие контрактур и деформаций, разработав программу растяжек и упражнений, где это необходимо.
- Предвидеть и свести к минимуму другие вторичные осложнения физического характера, рекомендуя фиксацию и долговечное медицинское оборудование. [40]
- Контролируйте функцию дыхания и дайте рекомендации по методам дыхательных упражнений и методам выведения выделений. [39]
- Ортопедические приспособления (такие как брекеты и инвалидные коляски) могут улучшить подвижность и способность к самообслуживанию. Облегающие съемные бандажи для ног, которые удерживают лодыжку на месте во время сна, могут отсрочить возникновение контрактур .
- По мере прогрессирования заболевания важна соответствующая респираторная поддержка.
- Сердечные проблемы могут потребовать установки кардиостимулятора . [41]
Препарат этеплирсен , антисмысловой олигоморфин , был одобрен в США для лечения мутаций, приводящих к пропуску экзона 51 дистрофина. Одобрение США было спорным [42] поскольку этеплирсену не удалось доказать клиническую пользу; [43] Европейское агентство по лекарственным средствам отказало ему в одобрении. [44] [45]
Препарат Аталурен (Трансларна) разрешен к применению в Европейском Союзе. [46] [47]
Антисмысловой олигонуклеотид голодирсен (Vyondys 53) был одобрен для медицинского использования в США в 2019 году для лечения случаев, когда может быть полезен пропуск экзона 53 транскрипта дистрофина. [48] [49]
Морфолино - антисмысловой олигонуклеотид вильтоларсен (Вилтепсо) был одобрен для медицинского использования в США в августе 2020 года для лечения мышечной дистрофии Дюшенна (МДД) у людей с подтвержденной мутацией гена МДД, поддающейся пропуску экзона 53. [50] Это второе одобренное таргетное лечение для людей с этим типом мутации в США. [50] Примерно 8% людей с МДД имеют мутацию, поддающуюся пропуску экзона 53. [50]
Казимерсен (Amondys 45) был одобрен для медицинского применения в США в феврале 2021 года. [51] и это первое одобренное FDA таргетное лечение для людей с подтвержденной мутацией гена мышечной дистрофии Дюшенна, поддающейся пропуску экзона 45. [51]
Комплексные междисциплинарные рекомендации по лечению мышечной дистрофии Дюшенна были разработаны Центрами США по контролю и профилактике заболеваний и опубликованы в 2010 году. [29] Обновление было опубликовано в 2018 году. [52] [53]
Деландистроген моксепарвовек США (Элевидис) — это препарат генной терапии, который в июне 2023 года получил ускоренное одобрение FDA для лечения четырех- и пятилетних детей. [54] [55]
США В марте 2024 года Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) одобрило пероральный препарат гивиностат (Дувизат) для лечения мышечной дистрофии Дюшенна у людей в возрасте шести лет и старше. Гивиностат — первый нестероидный препарат , получивший одобрение FDA для лечения всех генетических вариантов мышечной дистрофии Дюшенна. Действуя как ингибитор гистондеацетилазы ( HDAC ), гивиностат воздействует на патогенные процессы в организме, что в конечном итоге приводит к уменьшению воспаления и потери мышечной массы, связанных с заболеванием. [56]
Прогноз
[ редактировать ]Мышечная дистрофия Дюшенна — редкое прогрессирующее заболевание, которое в конечном итоге поражает все произвольные мышцы, а на более поздних стадиях вовлекает в процесс сердце и дыхательные мышцы. Ожидаемая продолжительность жизни оценивается примерно в 25–26 лет. [18] [57] но это варьируется. Средняя продолжительность жизни людей, родившихся с мышечной дистрофией Дюшенна после 1990 года, составляет примерно 28–30 лет. [11] [10] При превосходном медицинском обслуживании пострадавшие мужчины часто доживают до 30 лет. [58] Дэвид Хэтч из Парижа, штат Мэн , возможно, был самым старым человеком в мире, заболевшим этим заболеванием; он дожил до 56 лет. [59] [60]
Наиболее распространенной непосредственной причиной смерти людей с мышечной дистрофией Дюшенна является дыхательная недостаточность . Осложнения лечения, такие как процедуры искусственной вентиляции легких и трахеотомии , также вызывают беспокойство. Следующей по значимости причиной смерти являются сердечно-сосудистые заболевания, такие как сердечная недостаточность, вызванная дилатационной кардиомиопатией . При респираторной поддержке средний возраст выживания может достигать 40 лет. В редких случаях люди с мышечной дистрофией Дюшенна доживали до сорока или пятидесяти лет при правильном положении в инвалидных колясках и кроватях и использовании искусственной вентиляции легких (ИВЛ). через трахеостому или мундштук), очистку дыхательных путей и сердечные препараты. [61] Раннее планирование необходимой поддержки для ухода в более позднем возрасте показало большую продолжительность жизни людей с мышечной дистрофией Дюшенна. [62]
Любопытно, что в модели мышечной дистрофии Дюшенна на мышах mdx недостаток дистрофина связан с повышенным уровнем кальция и мионекрозом скелетных мышц. Внутренние мышцы гортани (ILM) защищены и не подвергаются мионекрозу. [63] ILM имеют профиль системы регуляции кальция, что позволяет предположить лучшую способность справляться с изменениями кальция по сравнению с другими мышцами, и это может дать механистическое понимание их уникальных патофизиологических свойств. [64] Кроме того, у пациентов с мышечной дистрофией Дюшенна также наблюдается повышенный уровень липопротеинов в плазме, что указывает на первичное состояние дислипидемии у пациентов. [65]
Эпидемиология
[ редактировать ]Мышечная дистрофия Дюшенна — наиболее распространенный тип мышечной дистрофии; при рождении он поражает примерно одного из 5000 мужчин. [3] Мышечная дистрофия Дюшенна встречается у одного из 3600 младенцев мужского пола. [18]
В США исследование 2010 года показало большее количество латиноамериканцев с мышечной дистрофией Дюшенна в возрасте от 5 до 54 лет по сравнению с неиспаноязычными белыми и неиспаноязычными чернокожими. [66] [67]
История
[ редактировать ]
Заболевание впервые описали неаполитанский врач Джованни Семмола в 1834 году и Гаэтано Конте в 1836 году. [68] [69] [70] Однако мышечная дистрофия Дюшенна названа в честь французского невролога Гийома-Бенжамена-Амана Дюшенна (1806–1875), который в издании 1861 года своей книги «Гипертрофический параплегии детского возраста» описал и подробно описал случай мальчика, который было такое состояние. Год спустя он представил фотографии своего пациента в своем «Альбоме патологических фотографий» . В 1868 году он рассказал о еще 13 пострадавших детях. Дюшенн был первым, кто провел биопсию ткани живого пациента для микроскопического исследования. [71] [72]
Общество и культура
[ редактировать ]Известные случаи
[ редактировать ]- Альфредо Феррари был итальянским автомобильным инженером, старшим сыном автопроизводителя Энцо Феррари и предполагаемым наследником компании по производству спортивных автомобилей своего отца Ferrari . Альфредо умер от МДД 30 июня 1956 года в возрасте 24 лет. [73] [74]
- Рэпер и защитник прав инвалидов Дариус Уимс страдал этим заболеванием и использовал свою известность для повышения осведомленности и финансирования лечения, как показано в документальном фильме «Дариус идет на запад» (2007). [75] Он умер в возрасте 27 лет в 2016 году. [76]
- Джонатана Эвисона В романе «Пересмотренные основы ухода» , опубликованном в 2012 году, изображен молодой человек, страдающий этой болезнью. В 2016 году Netflix выпустил фильм «Основы заботы » по мотивам романа. [77]
Исследовать
[ редактировать ]Этот раздел необходимо обновить . ( август 2019 г. ) |
Продолжаются попытки найти лекарства, которые возвращают способность вырабатывать дистрофин или атрофин. [78] Другие усилия включают попытки заблокировать проникновение ионов кальция в мышечные клетки. [79]
пропуск экзонов
[ редактировать ]Антисмысловые олигонуклеотиды (олигонуклеотиды), структурные аналоги ДНК, являются основой потенциального лечения 10% людей с мышечной дистрофией Дюшенна. [80] Соединения позволяют пропускать дефектные части гена дистрофина при его транскрипции в РНК для производства белка, что позволяет производить все еще усеченную, но более функциональную версию белка. [81] Она также известна как терапия подавления нонсенса. [82]
Два типа антисмысловых олигонуклеотидов, 2'-O-метилфосфоротиоатные олигонуклеотиды (например, дрисаперсен ) и морфолиноолигонуклеотиды (например, этеплирсен ), имеют предварительные доказательства полезности и изучаются. [83] Целью Этеплирсена является пропуск экзона 51. [83] «Например, пропуск экзона 51 восстанавливает рамку считывания примерно у 15% всех мальчиков с делециями. Было высказано предположение, что, имея 10 АОН для пропуска 10 разных экзонов, можно было бы справиться с более чем 70% всех Мальчики с МДД с делециями». [80] Это составляет около 1,5% случаев. [80]

Люди с мышечной дистрофией Беккера , которая протекает легче, чем МДД, имеют форму дистрофина, которая функциональна, хотя она короче нормального дистрофина. [84] В 1990 году Англия и др. заметили, что у пациента с легкой мышечной дистрофией Беккера не хватало 46% области, кодирующей дистрофин. [84] Эта функциональная, но усеченная форма дистрофина породила представление о том, что более короткий дистрофин все еще может быть терапевтически полезным. Одновременно Коле и др. модифицировали сплайсинг путем нацеливания на пре-мРНК антисмысловых олигонуклеотидов (АОН). [85] Коле продемонстрировал успех в использовании AON, нацеленных на сплайсинг, для исправления неправильного сплайсинга в клетках, удаленных от пациентов с бета-талассемией. [86] [87] Группа Уилтона проверила пропуск экзонов при мышечной дистрофии. [88] [89]
Генная терапия
[ редактировать ]Исследователи работают над методом редактирования генов, чтобы исправить мутацию, приводящую к мышечной дистрофии Дюшенна (МДД). [90] Исследователи использовали метод под названием CRISPR/Cas9-опосредованное редактирование генома , который может точно удалить мутацию гена дистрофина организма в ДНК, позволяя механизмам восстановления ДНК заменить его нормальной копией гена. [91] [92]
Редактирование генома человека с помощью системы CRISPR/Cas9 в настоящее время невозможно. Тем не менее, благодаря развитию технологий, возможно, в будущем можно будет использовать этот метод для разработки методов лечения МДД. [93] [94] В 2007 году исследователи провели первое в мире клиническое (вирусно-опосредованное) исследование генной терапии болезни Дюшенна. [95]
Биострофин является вектором доставки генной терапии при лечении мышечной дистрофии Дюшенна и Беккера мышечной дистрофии . [96]
Будущие разработки
[ редактировать ]Несколько лекарств, предназначенных для устранения основной причины, находятся в стадии разработки, включая генную терапию и антисмысловые препараты . [3] Другие используемые лекарства включают кортикостероиды для замедления дегенерации мышц. [2] Физиотерапия , ортопедические брекеты и корректирующая хирургия могут помочь при некоторых симптомах. [2] в то время как вспомогательная вентиляция может потребоваться людям со слабостью дыхательных мышц . [3] Результаты зависят от конкретного типа расстройства. [97] [3]
Ссылки
[ редактировать ]- ^ «Дюшенн» . Словарь Merriam-Webster.com .
- ^ Jump up to: а б с д и ж г час я дж «Информационная страница NINDS о мышечной дистрофии» . Национальный институт неврологических расстройств и инсульта (NINDS) . 4 марта 2016 г. Архивировано из оригинала 30 июля 2016 г. . Проверено 12 сентября 2016 г.
 В данную статью включен текст из этого источника, находящегося в свободном доступе .
В данную статью включен текст из этого источника, находящегося в свободном доступе . - ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С аа «Мышечная дистрофия: надежда через исследования» . Национальный институт неврологических расстройств и инсульта (NINDS) . 4 марта 2016 г. Архивировано из оригинала 30 сентября 2016 г. Проверено 12 сентября 2016 г. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ «Мышечная дистрофия» . Национальный институт неврологических расстройств и инсульта (NINDS) . 30 октября 2023 года. Архивировано из оригинала 31 марта 2024 года . Проверено 31 марта 2024 г. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ Jump up to: а б с Нозоэ К.Т., Акамин Р.Т., Маццотти Д.Р., Полесел Д.Н., Гроссклаусс Л.Ф., Туфик С. и др. (2016). «Фенотипические контрасты мышечной дистрофии Дюшенна у женщин: два описания случаев» . Наука сна . 9 (3): 129–133. дои : 10.1016/j.slsci.2016.07.004 . ПМК 5241604 . ПМИД 28123647 .
- ^ «Мышечная дистрофия: надежда через исследования» . Национальный институт неврологических расстройств и инсульта (NINDS) . Сентябрь 2013. Архивировано из оригинала 31 марта 2024 года . Проверено 31 марта 2024 г. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ https://catalog.ninds.nih.gov/sites/default/files/publications/muscular-dystrophy-hope-through-research.pdf. Архивировано 31 марта 2024 г. в Wayback Machine. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ Крисафулли С., Султана Дж., Фонтана А., Сальво Ф., Мессина С., Трифиро Дж. (июнь 2020 г.). «Глобальная эпидемиология мышечной дистрофии Дюшенна: обновленный систематический обзор и метаанализ» . Сиротский журнал редких заболеваний . 15 (1): 141. дои : 10.1186/s13023-020-01430-8 . ПМЦ 7275323 . ПМИД 32503598 .
- ^ «Мышечная дистрофия Дюшенна (МДД)» . Ассоциация мышечной дистрофии . 17 ноября 2017 года. Архивировано из оригинала 15 ноября 2022 года . Проверено 15 ноября 2022 г.
- ^ Jump up to: а б Ландфельдт Э., Томпсон Р., Сейерсен Т., Макмиллан Х.Дж., Киршнер Дж., Лохмюллер Х. (2020). «Ожидаемая продолжительность жизни при рождении при мышечной дистрофии Дюшенна: систематический обзор и метаанализ» . Европейский журнал эпидемиологии . 35 (7): 643–653. дои : 10.1007/s10654-020-00613-8 . ISSN 1573-7284 . ПМЦ 7387367 . ПМИД 32107739 .
- ^ Jump up to: а б Брумфилд Дж., Хилл М., Гульери М., Кроутер М., Абрамс К. (7 декабря 2021 г.). «Продолжительность жизни при мышечной дистрофии Дюшенна: метаанализ воспроизведенных индивидуальных данных пациентов» . Неврология . 97 (23): e2304–e2314. дои : 10.1212/WNL.0000000000012910 . ISSN 0028-3878 . ПМЦ 8665435 . PMID 34645707 .
- ^ Jump up to: а б с д и «Мышечная дистрофия Дюшенна» . Информационный центр по генетическим и редким заболеваниям (GARD) . Архивировано из оригинала 23 ноября 2016 года . Проверено 24 января 2021 г. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ Анджелини С., Таска Э. (декабрь 2012 г.). «Утомляемость при мышечных дистрофиях» . Нервно-мышечные расстройства . 22 Приложение 3 (3): S214–S220. дои : 10.1016/j.nmd.2012.10.010 . ПМЦ 3526799 . ПМИД 23182642 .
- ^ Jump up to: а б Роуленд LP (1985). «Клиническая перспектива: фенотипическое выражение при мышечной дистрофии» . В Строман С., Вольф С. (ред.). Экспрессия генов в мышцах . Достижения экспериментальной медицины и биологии. Пленум Пресс. стр. 3–5. ISBN 978-1-4684-4907-5 . Архивировано из оригинала 29 сентября 2020 года . Проверено 7 августа 2019 г.
- ^ Jump up to: а б с д и ж Даррас Б.Т., Урион Д.К., Гош П.С. (2018). «Дистрофинопатии». Джин Обзоры . Сиэтл (Вашингтон): Вашингтонский университет. ПМИД 20301298 .
- ^ Эмери А.Е., Мунтони Ф., Куинливан Р. (2015). Мышечная дистрофия Дюшенна (Четвертое изд.). ОУП Оксфорд. ISBN 978-0-19968148-8 . Архивировано из оригинала 31 марта 2024 года . Проверено 27 мая 2020 г.
- ^ Jump up to: а б «Мышечная дистрофия – Симптомы и причины» . Клиника Мэйо . Архивировано из оригинала 6 февраля 2015 года . Проверено 6 февраля 2015 г.
- ^ Jump up to: а б с д Энциклопедия MedlinePlus : мышечная дистрофия Дюшенна
- ^ Сазерленд Д.Х., Олшен Р., Купер Л., Вятт М., Лич Дж., Мубарак С. и др. (февраль 1981 г.). «Патомеханика походки при мышечной дистрофии Дюшенна». Медицина развития и детская неврология . 23 (1): 3–22. дои : 10.1111/j.1469-8749.1981.tb08442.x . ПМИД 7202868 . S2CID 895379 .
- ^ Баптиста ЧР, Коста А.А., Пиццато ТМ, Соуза ФБ, Маттиелло-Сверзут AC (2014). «Выравнивание осанки у детей с мышечной дистрофией Дюшенна и ее связь с балансом» . Бразильский журнал физиотерапии . 18 (2): 119–126. дои : 10.1590/s1413-35552012005000152 . ПМЦ 4183248 . ПМИД 24838810 .
- ^ Дооренверд Н., Махфуз А., ван Путтен М., Калияперумал Р., Т' Хоен П.А., Хендриксен Дж.Г. и др. (октябрь 2017 г.). «Время и локализация экспрессии изоформы дистрофина человека дают представление о когнитивном фенотипе мышечной дистрофии Дюшенна» . Научные отчеты . 7 (1): 12575. Бибкод : 2017NatSR...712575D . дои : 10.1038/s41598-017-12981-5 . ПМЦ 5626779 . ПМИД 28974727 .
- ^ Интернет-менделевское наследование у человека (OMIM): мышечная дистрофия, тип Дюшенна; ДМД - 310200
- ^ Вера CD, Чжан А, Панг П.Д., Ву Дж.К. (2022). «Лечение мышечной дистрофии Дюшенна: перспективы стволовых клеток, искусственного интеллекта и мультиомиков» . Границы сердечно-сосудистой медицины . 9 : 851491. дои : 10.3389/fcvm.2022.851491 . ПМК 8960141 . ПМИД 35360042 .
- ^ «Мышечная дистрофия Дюшенна: патофизиологические последствия передачи сигналов митохондриального кальция и производства АФК» . 2 мая 2012 года. Архивировано из оригинала 2 мая 2012 года . Проверено 29 июня 2014 г.
- ^ Валь М. (21 октября 2016 г.). «Квест - Статья - Но у девушек не бывает Дюшенна, или нет? - Квестовая статья» . Ассоциация мышечной дистрофии . Архивировано из оригинала 12 апреля 2019 года . Проверено 6 июля 2019 г.
- ^ Вучерпфенниг Дж (6 октября 2016 г.). «Если у мужчины мышечная дистрофия Дюшенна, каковы шансы, что у его детей будет МДД?» . Технический интерактив . Спросите генетика . Проверено 5 августа 2024 г.
- ^ Нико Б., Рибатти Д. (январь 2012 г.). «Морфофункциональные аспекты гематоэнцефалического барьера». Современный метаболизм лекарств . 13 (1): 50–60. дои : 10.2174/138920012798356970 . ПМИД 22292807 .
- ^ «Университет Юты по мышечной дистрофии» . Геном.utah.edu. 28 ноября 2009 г. Архивировано из оригинала 14 сентября 2003 г. Проверено 16 февраля 2013 г.
- ^ Jump up to: а б Бушби К., Финкель Р., Бирнкрант Д.Д., Кейс Л.Е., Клеменс П.Р., Крайп Л. и др. (январь 2010 г.). «Диагностика и лечение мышечной дистрофии Дюшенна, часть 1: диагностика, фармакологическое и психосоциальное лечение». «Ланцет». Неврология . 9 (1): 77–93. CiteSeerX 10.1.1.176.4466 . дои : 10.1016/s1474-4422(09)70271-6 . ПМИД 19945913 . S2CID 328499 .
- ^ Николсон Л.В., Джонсон М.А., Бушби К.М., Гарднер-Медвин Д., Кертис А., Гинджар И.Б. и др. (сентябрь 1993 г.). «Комплексное исследование 100 пациентов с мышечной дистрофией, связанной с Xp21, с использованием клинических, генетических, иммунохимических и гистопатологических данных. Часть 2. Корреляции внутри отдельных пациентов» . Журнал медицинской генетики . 30 (9): 737–744. дои : 10.1136/jmg.30.9.737 . ПМК 1016530 . ПМИД 8411068 .
- ^ Мунтони Ф (август 2001 г.). «Действительно ли необходима мышечная биопсия при дистрофии Дюшенна?». Неврология . 57 (4): 574–575. дои : 10.1212/wnl.57.4.574 . ПМИД 11524463 . S2CID 13474827 .
- ^ Фланиган К.М., фон Нидерхаузерн А., Данн Д.М., Олдер Дж., Менделл Дж.Р., Вайс Р.Б. (апрель 2003 г.). «Быстрый анализ прямой последовательности гена дистрофина» . Американский журнал генетики человека . 72 (4): 931–939. дои : 10.1086/374176 . ПМК 1180355 . ПМИД 12632325 .
- ^ Бексач М.С., Танакан А., Айдын Хакли Д., Оргул Г., Сояк Б., Балчи Хайта Б. и др. (30 июля 2018 г.). «Гестационные исходы у беременных женщин, прошедших инвазивное пренатальное тестирование для пренатальной диагностики мышечной дистрофии Дюшенна» . Журнал беременности . 2018 : 9718316. doi : 10.1155/2018/9718316 . ПМК 6091284 . ПМИД 30151283 .
- ^ Девани С.А., Паломаки Г.Е., Скотт Дж.А., Бьянки Д.В. (август 2011 г.). «Неинвазивное определение пола плода с использованием бесклеточной ДНК плода: систематический обзор и метаанализ» . ДЖАМА . 306 (6): 627–636. дои : 10.1001/jama.2011.1114 . ПМК 4526182 . ПМИД 21828326 .
- ^ «Заявление о мышечной дистрофии Дюшенна» . США Управление по контролю за продуктами и лекарствами (FDA). 31 октября 2014 г. Архивировано из оригинала 2 ноября 2014 г. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ Дэни А., Барб С., Рапин А., Ревейер С., Хардуэн Ж.Б., Моррон И. и др. (ноябрь 2015 г.). «Построение опросника качества жизни при медленно прогрессирующих нервно-мышечных заболеваниях». Исследование качества жизни . 24 (11): 2615–2623. дои : 10.1007/s11136-015-1013-8 . ПМИД 26141500 . S2CID 25834947 .
- ^ Фальзарано М.С., Скоттон С., Пассарелли С., Ферлини А. (октябрь 2015 г.). «Мышечная дистрофия Дюшенна: от диагностики к терапии» . Молекулы . 20 (10): 18168–18184. дои : 10.3390/molecules201018168 . ПМК 6332113 . ПМИД 26457695 .
- ^ Мэтьюз Э., Брассингтон Р., Кунцер Т., Джичи Ф., Манзур А.Ю. (май 2016 г.). «Кортикостероиды для лечения мышечной дистрофии Дюшенна» . Кокрановская база данных систематических обзоров . 5 (5): CD003725. дои : 10.1002/14651858.CD003725.pub4 . ПМЦ 8580515 . ПМИД 27149418 .
- ^ Jump up to: а б «Мышечная дистрофия Дюшенна» . Физиопедия . Архивировано из оригинала 10 октября 2022 года . Проверено 10 октября 2022 г.
- ^ Педлоу К., Макдоно С., Леннон С., Керр С., Брэдбери I (октябрь 2019 г.). «Помощь в стоянии при мышечной дистрофии Дюшенна» . Кокрановская база данных систематических обзоров . 10 (10): CD011550. дои : 10.1002/14651858.CD011550.pub2 . ПМК 6790222 . ПМИД 31606891 .
- ^ Верхарт Д., Ричардс К., Рафаэль-Фортни Дж.А., Раман С.В. (январь 2011 г.). «Поражение сердца у пациентов с мышечными дистрофиями: фенотип магнитно-резонансной томографии и генотипические соображения» . Кровообращение: сердечно-сосудистая визуализация . 4 (1): 67–76. дои : 10.1161/CIRCIMAGING.110.960740 . ПМК 3057042 . ПМИД 21245364 .
- ^ «Железнодорожное сообщение в FDA» . Природная биотехнология . 34 (11): 1078. Ноябрь 2016 г. doi : 10.1038/nbt.3733 . ПМИД 27824847 .
- ^ «FDA предоставляет ускоренное одобрение первому препарату для лечения мышечной дистрофии Дюшенна» (пресс-релиз). США Управление по контролю за продуктами и лекарствами (FDA). 19 сентября 2016 г. Архивировано из оригинала 2 августа 2019 г. . Проверено 8 июля 2019 г. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ «CHMP не рекомендует использовать Этеплирсен в лечении DMD» . Медскейп . Архивировано из оригинала 9 июля 2019 года . Проверено 9 июля 2019 г.
- ^ «Экзондис» . Европейское агентство по лекарственным средствам . 17 сентября 2018 г. Архивировано из оригинала 26 ноября 2020 г. . Проверено 3 декабря 2022 г.
- ^ «Трансларна ЭПАР» . Европейское агентство лекарственных средств (EMA) . Архивировано из оригинала 29 октября 2020 года . Проверено 14 августа 2020 г.
- ^ «Трансларна – Сводная характеристика продукции (СмПК)» . (эмс) . 24 апреля 2017 года. Архивировано из оригинала 15 июля 2017 года . Проверено 18 июня 2017 г.
- ^ «FDA предоставляет ускоренное одобрение первому таргетному лечению редкой мутации мышечной дистрофии Дюшенна» (пресс-релиз). США Управление по контролю за продуктами и лекарствами (FDA). 12 декабря 2019 года. Архивировано из оригинала 13 декабря 2019 года . Проверено 12 декабря 2019 г. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ «Пакет одобрения препарата: Виондис 53 (голодирсен)» . США Управление по контролю за продуктами и лекарствами (FDA). 21 января 2020 года. Архивировано из оригинала 2 марта 2020 года . Проверено 22 января 2020 г.
- ^ Jump up to: а б с «FDA одобрило таргетное лечение редкой мутации мышечной дистрофии Дюшенна» (пресс-релиз). США Управление по контролю за продуктами и лекарствами (FDA). 12 августа 2020 года. Архивировано из оригинала 20 августа 2020 года . Проверено 12 августа 2020 г. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ Jump up to: а б «FDA одобрило таргетное лечение редкой мутации мышечной дистрофии Дюшенна» (пресс-релиз). США Управление по контролю за продуктами и лекарствами (FDA). 25 февраля 2021 года. Архивировано из оригинала 3 августа 2021 года . Проверено 25 февраля 2021 г. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ Бирнкрант Д.Д., Бушби К., Банн К.М., Апкон С.Д., Блэквелл А., Брамбо Д. и др. (март 2018 г.). «Диагностика и лечение мышечной дистрофии Дюшенна, часть 1: диагностика, нервно-мышечная, реабилитация, эндокринная, желудочно-кишечная и диетологическая помощь» . «Ланцет». Неврология . 17 (3): 251–267. дои : 10.1016/S1474-4422(18)30024-3 . ПМК 5869704 . ПМИД 29395989 .
- ^ Бирнкрант Д.Д., Бушби К., Банн К.М., Алман Б.А., Апкон С.Д., Блэквелл А. и др. (апрель 2018 г.). «Диагностика и лечение мышечной дистрофии Дюшенна, часть 2: респираторное, сердечное, здоровье костей и ортопедическое лечение» . «Ланцет». Неврология . 17 (4): 347–361. дои : 10.1016/S1474-4422(18)30025-5 . ПМК 5889091 . ПМИД 29395990 .
- ^ «FDA одобрило первую генную терапию для лечения некоторых пациентов с мышечной дистрофией Дюшенна» (пресс-релиз). США Управление по контролю за продуктами и лекарствами (FDA). 22 июня 2023 года. Архивировано из оригинала 29 ноября 2023 года . Проверено 22 июня 2023 г. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ «Sarepta Therapeutics объявляет об одобрении FDA препарата Элевидис, первого генного препарата для лечения мышечной дистрофии Дюшенна» (пресс-релиз). Сарепта Терапевтикс. 22 июня 2023 года. Архивировано из оригинала 23 июня 2023 года . Проверено 22 июня 2023 г. - через Business Wire.
- ^ «FDA одобрило нестероидное лечение мышечной дистрофии Дюшенна» . США Управление по контролю за продуктами и лекарствами (FDA) (пресс-релиз). 21 марта 2024 года. Архивировано из оригинала 23 марта 2024 года . Проверено 23 марта 2024 г. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ Лисак Р.П., Труонг Д.Д., Кэрролл В., Бхидаясири Р. (2011). Международная неврология . Уайли. п. 222. ИСБН 978-1-4443-1701-5 .
- ^ «Мышечная дистрофия Дюшенна (МДД) | Кампания по борьбе с мышечной дистрофией» . Muscular-dystrophy.org. Архивировано из оригинала 21 января 2013 года . Проверено 16 февраля 2013 г.
- ^ Картер Н. (14 января 2021 г.). «Жительница дома престарелых бросает вызов COVID и снова хочет поесть вне дома» . Солнечный журнал . Архивировано из оригинала 14 января 2021 года . Проверено 14 января 2021 г.
- ^ «Некрологи - Кремации и похороны Клиффа Грея» . Февраль 2022 года. Архивировано из оригинала 28 декабря 2022 года . Проверено 28 декабря 2022 г.
- ^ Кьени П., Шолле С., Делаланд П., Ле Фор М., Маго А., Переон Ю. и др. (сентябрь 2013 г.). «Эволюция ожидаемой продолжительности жизни пациентов с мышечной дистрофией Дюшенна в центре AFM Йолен де Кеппер в период с 1981 по 2011 год» . Анналы физической и реабилитационной медицины . 56 (6): 443–454. дои : 10.1016/j.rehab.2013.06.002 . ПМИД 23876223 .
- ^ Краина А., Подрабски П., Стейнхарт Л., Эндрис Дж., Куфаль Л. (22 ноября 2012 г.). «[Личный экспериментальный опыт введения жидких облитерирующих средств с использованием чрескожных внутриартериальных баллонных катетеров с контролируемой утечкой]» . Сборник научных трудов медицинского факультета Карлова университета в Градце-Кралове. Добавка . 30 (2): 201–211. дои : 10.1186/1750-1172-7-S2-A8 . ПМЦ 3504593 . ПМИД 3504593 .
- ^ Маркес М.Дж., Ферретти Р., Вомеро В.У., Минатель Э., Нето Х.С. (март 2007 г.). «Внутренние мышцы гортани защищены от мионекроза в модели мышечной дистрофии Дюшенна на мышах mdx». Мышцы и нервы . 35 (3): 349–353. дои : 10.1002/mus.20697 . ПМИД 17143878 . S2CID 41968787 .
- ^ Ферретти Р., Маркес М.Дж., Хурана Т.С., Санто Нето Х. (июнь 2015 г.). «Экспрессия кальций-буферных белков во внутренних мышцах гортани крысы» . Физиологические отчеты . 3 (6): е12409. дои : 10.14814/phy2.12409 . ПМК 4510619 . ПМИД 26109185 .
- ^ Уайт З., Хаким С.Х., Терет М., Ян Н.Н., Росси Ф., Кокс Д. и др. (июль 2020 г.). «Высокая распространенность аномалий липидов плазмы при мышечных дистрофиях Дюшенна и Беккера у людей и собак свидетельствует о новом типе первичной генетической дислипидемии» . Журнал клинической липидологии . 14 (4): 459–469.e0. doi : 10.1016/j.jacl.2020.05.098 . ПМЦ 7492428 . ПМИД 32593511 . S2CID 219741257 .
- ^ «Популяционная распространенность мышечных дистрофий Дюшенна и Беккера в Соединенных Штатах» . Центры США по контролю и профилактике заболеваний (CDC). 5 января 2018 г. Архивировано из оригинала 18 ноября 2018 г. Проверено 18 ноября 2018 г.
- ^ Ромитти П.А., Чжу Ю., Пужанкара С., Джеймс К.А., Набукера С.К., Замба Г.К. и др. (март 2015 г.). «Распространенность мышечных дистрофий Дюшенна и Беккера в Соединенных Штатах» . Педиатрия . 135 (3): 513–21. дои : 10.1542/пед.2014-2044 . ПМЦ 4477633 . ПМИД 25687144 .
- ^ Политано Л. «Кардиомиология и медицинская генетика» (на итальянском языке). Второй университет Неаполя . Архивировано из оригинала 4 июля 2015 года . Проверено 24 августа 2015 г.
- ^ Де Роза Дж. (октябрь 2005 г.). «От Конте до Дюшенна» [Конте в Дюшенне]. ДМ (на итальянском языке). Итальянский союз борьбы с мышечной дистрофией. Архивировано из оригинала 4 марта 2016 года . Проверено 24 августа 2015 г.
- ^ Нигро Дж (декабрь 2010 г.). «Сто семьдесят пять лет неаполитанского вклада в борьбу с мышечными заболеваниями» . Акта Миологика . 29 (3): 369–391. ПМК 3146338 . ПМИД 21574522 .
- ^ «Мышечная дистрофия Дюшенна» . Medterms.com. 27 апреля 2011 года. Архивировано из оригинала 6 августа 2012 года . Проверено 16 февраля 2013 г.
- ^ Дюшенн де Булонь в журнале «Кто это назвал?»
- ^ Сюзанна Ким (21 октября 2015 г.). «Что вы не знали о семье Феррари» . Новости Эй-Би-Си. Архивировано из оригинала 31 августа 2023 года . Проверено 31 августа 2023 г.
- ^ Команда GearShifters (13 сентября 2022 г.). «Как умер Дино Феррари?» . Переключатели передач. Архивировано из оригинала 31 августа 2023 года . Проверено 31 августа 2023 г.
- ^ Макфадден С., Джонсон Э., Эффрон Л. (22 ноября 2012 г.). «Следующая глава Дариуса Уимса: рэп-звезда с мышечной дистрофией Дюшенна проходит клиническое исследование» . Новости АВС . Архивировано из оригинала 5 августа 2016 года . Проверено 29 июня 2016 г.
- ^ Эрик Джонсон (10 октября 2016 г.). «Активист по правам инвалидов Дариус Уимс проигрывает битву с мышечной дистрофией Дюшенна» . Новости Эй-Би-Си. Архивировано из оригинала 31 августа 2023 года . Проверено 31 августа 2023 г.
- ^ Беркшир G (23 января 2016 г.). «Обзор фильма «Сандэнс»: «Основы заботы» » . Разнообразие . Архивировано из оригинала 21 октября 2021 года . Проверено 21 октября 2021 г.
- ^ Гиро С., Дэвис К.Э. (июнь 2017 г.). «Фармакологические достижения в лечении мышечной дистрофии Дюшенна» . Современное мнение в фармакологии . 34 : 36–48. дои : 10.1016/j.coph.2017.04.002 . ПМИД 28486179 .
- ^ Аллен Д.Г., Джервасио О.Л., Йенг Э.В., Уайтхед Н.П. (февраль 2010 г.). «Кальций и пути повреждения при мышечной дистрофии». Канадский журнал физиологии и фармакологии . 88 (2): 83–91. дои : 10.1139/Y09-058 . ПМИД 20237582 .
- ^ Jump up to: а б с Ското М., Финкель Р., Меркури Э., Мунтони Ф. (август 2018 г.). «Генетическая терапия наследственных нервно-мышечных заболеваний» . «Ланцет». Здоровье детей и подростков . 2 (8): 600–609. дои : 10.1016/S2352-4642(18)30140-8 . ПМИД 30119719 . S2CID 52032568 . Архивировано из оригинала 17 августа 2021 года . Проверено 16 ноября 2022 г.
- ^ Данкли М.Г., Манохаран М., Виллиет П., Эперон И.С., Диксон Дж. (июль 1998 г.). «Модификация сплайсинга гена дистрофина в культивируемых мышечных клетках Mdx антисмысловыми олигорибонуклеотидами» . Молекулярная генетика человека . 7 (7): 1083–1090. дои : 10.1093/hmg/7.7.1083 . ПМИД 9618164 .
- ^ Финкель Р.С. (сентябрь 2010 г.). «Стратегии чтения для подавления нонсенс-мутаций при мышечной дистрофии Дюшенна/Беккера: аминогликозиды и аталурен (PTC124)» . Журнал детской неврологии . 25 (9): 1158–1164. дои : 10.1177/0883073810371129 . ПМЦ 3674569 . ПМИД 20519671 .
- ^ Jump up to: а б «FDA предоставляет ускоренное одобрение первому препарату для лечения мышечной дистрофии Дюшенна» (пресс-релиз). США Управление по контролю за продуктами и лекарствами (FDA). 19 сентября 2016 года. Архивировано из оригинала 11 декабря 2016 года . Проверено 12 декабря 2016 г. В данную статью включен текст из этого источника, находящегося в свободном доступе .
- ^ Jump up to: а б Англия С.Б., Николсон Л.В., Джонсон М.А., Форрест С.М., Лав Д.Р., Зубжицка-Гаарн Э.Э. и др. (январь 1990 г.). «Очень легкая мышечная дистрофия, связанная с удалением 46% дистрофина». Природа . 343 (6254): 180–182. Бибкод : 1990Natur.343..180E . дои : 10.1038/343180a0 . ПМИД 2404210 . S2CID 4349360 .
- ^ Домински З., Коле Р. (сентябрь 1993 г.). «Восстановление правильного сплайсинга талассемической пре-мРНК антисмысловыми олигонуклеотидами» . Труды Национальной академии наук Соединенных Штатов Америки . 90 (18): 8673–8677. Бибкод : 1993PNAS...90.8673D . дои : 10.1073/pnas.90.18.8673 . ПМК 47420 . ПМИД 8378346 .
- ^ Ласерра Г., Сераковска Х., Карестия С., Фучароен С., Саммертон Дж., Веллер Д. и др. (август 2000 г.). «Восстановление синтеза гемоглобина А в эритроидных клетках периферической крови больных талассемией» . Труды Национальной академии наук Соединенных Штатов Америки . 97 (17): 9591–9596. Бибкод : 2000PNAS...97.9591L . дои : 10.1073/pnas.97.17.9591 . ПМК 16909 . ПМИД 10944225 .
- ^ Суванмани Т., Сераковска Х., Ласерра Г., Свасти С., Кирби С., Уолш С.Э. и др. (сентябрь 2002 г.). «Восстановление экспрессии гена бета-глобина человека в талассемических эритроидных клетках мыши и человека IVS2-654 путем свободного поглощения антисмысловых олигонуклеотидов». Молекулярная фармакология . 62 (3): 545–553. дои : 10.1124/моль.62.3.545 . ПМИД 12181431 .
- ^ Уилтон С.Д., Ллойд Ф., Карвилл К., Флетчер С., Ханиман К., Агравал С. и др. (июль 1999 г.). «Специфическое удаление нонсенс-мутации из мРНК дистрофина mdx с использованием антисмысловых олигонуклеотидов». Нервно-мышечные расстройства . 9 (5): 330–338. дои : 10.1016/S0960-8966(99)00010-3 . ПМИД 10407856 . S2CID 20678312 .
- ^ Уилтон С.Д., Фолл А.М., Хардинг П.Л., МакКлори Г., Коулман С., Флетчер С. (июль 2007 г.). «Пропуск экзона, индуцированный антисмысловыми олигонуклеотидами, в транскрипте гена дистрофина человека» . Молекулярная терапия . 15 (7): 1288–1296. дои : 10.1038/sj.mt.6300095 . ПМИД 17285139 .
- ^ Лонг С., Ли Х., Тибурси М., Родригес-Кайседо С., Кириченко В., Чжоу Х. и др. (январь 2018 г.). «Коррекция разнообразных мутаций мышечной дистрофии в искусственно созданной сердечной мышце человека путем редактирования генома в одном месте» . Достижения науки . 4 (1): eaap9004. Бибкод : 2018SciA....4.9004L . дои : 10.1126/sciadv.aap9004 . ПМК 5796795 . ПМИД 29404407 .
- ^ Коэн Дж. (30 августа 2018 г.). «Редактирование генов собак дает надежду на лечение мышечной дистрофии человека». Наука . дои : 10.1126/science.aav2676 . S2CID 92204241 .
- ^ Патманатан С.Н., Гнанасегаран Н., Лим М.Н., Хусайни Р., Факируддин К.С., Закария З. (2018). «CRISPR/Cas9 в исследованиях стволовых клеток: текущее применение и перспективы на будущее». Современные исследования и терапия стволовыми клетками . 13 (8): 632–644. дои : 10.2174/1574888X13666180613081443 . ПМИД 29895256 . S2CID 48357156 .
- ^ Лонг С., Макэналли-младший, Шелтон Дж.М., Миро А.А., Бассель-Дюби Р., Олсон Э.Н. (сентябрь 2014 г.). «Профилактика мышечной дистрофии у мышей с помощью CRISPR/Cas9-опосредованного редактирования ДНК зародышевой линии» . Наука . 345 (6201): 1184–1188. Бибкод : 2014Sci...345.1184L . дои : 10.1126/science.1254445 . ПМК 4398027 . ПМИД 25123483 .
- ^ Уэйд Н. (31 декабря 2015 г.). «Редактирование генов дает надежду на лечение мышечной дистрофии Дюшенна, показывают исследования» . Нью-Йорк Таймс . Архивировано из оригинала 2 января 2016 года . Проверено 1 января 2016 г.
- ^ Родино-Клапак Л.Р., Чикойн Л.Г., Каспар Б.К., Менделл-младший (сентябрь 2007 г.). «Генная терапия мышечной дистрофии Дюшенна: ожидания и проблемы» . Архив неврологии . 64 (9): 1236–1241. дои : 10.1001/archneur.64.9.1236 . ПМИД 17846262 .
- ^ Хурдаян В.К., Боззо Дж., Проус Дж.Р. (октябрь 2005 г.). «Хроники открытия лекарств». Новости и перспективы наркотиков . 18 (8): 517–522. дои : 10.1358/dnp.2005.18.8.953409 . ПМИД 16391721 .
- ^ «Информационная страница NINDS о мышечной дистрофии» . НИНДС . 4 марта 2016 г. Архивировано из оригинала 30 июля 2016 г. . Проверено 12 сентября 2016 г.