Врожденная мышечная дистрофия
| Врожденная мышечная дистрофия | |
|---|---|
 | |
| Аутосомно-рецессивный тип наследования обычно является типом наследования ВМД. | |
| Специальность | Неврология |
| Симптомы | Мышечная слабость [ 1 ] |
| Типы | 17 типов КМД [ 1 ] |
| Метод диагностики | ЕДА, ЭМГ [ 2 ] |
| Уход | В настоящее время нет никакого лечения; следует контролировать функцию сердца и дыхания [ 3 ] |
Врожденные мышечные дистрофии — это аутосомно -рецессивно наследуемые заболевания мышц . Они представляют собой группу гетерогенных нарушений, характеризующихся мышечной слабостью, присутствующей при рождении, и различными изменениями при биопсии мышц , которые варьируются от миопатических до явно дистрофических в зависимости от возраста, в котором проводится биопсия. [ 1 ] [ 4 ]
Признаки и симптомы
[ редактировать ]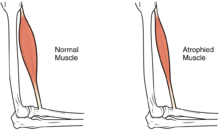
У большинства детей с ВМД наблюдается некоторая прогрессирующая мышечная слабость или атрофия мышц ( атрофия ), хотя степень и симптомы прогрессирования могут быть разной. Слабость обозначается как гипотония или отсутствие мышечного тонуса, из-за чего ребенок может казаться нестабильным. [ 1 ] [ 5 ] Со временем у большинства пациентов развиваются контрактуры суставов или фиксированные деформации суставов. [ 6 ]
У детей может быть медленная моторика ; например, переворачиваться, сидеть или ходить, или могут даже не достичь этих вех жизни. Некоторые из более редких форм CMD могут привести к серьезным нарушениям обучаемости. [ 7 ]
Генетика
[ редактировать ]Врожденные мышечные дистрофии (ВМД) наследуются аутосомно-рецессивно, за исключением некоторых случаев генной мутации de novo и врожденной мышечной дистрофии Ульриха. [ 8 ] [ 9 ] Это означает, что в большинстве случаев оба родителя должны быть носителями гена CMD, чтобы он передался по наследству. ВМД гетерогенны, и к настоящему времени обнаружено 35 генов, участвующих в различных формах ВМД, возникающих в результате этих мутаций. [ 10 ] [ 11 ] [ 12 ] [ 13 ] [ 8 ] Существуют различные формы ВМД, которые часто классифицируются по изменениям белка, вызванным атипичным геном.
Одна группа форм - это те, при которых у пациента с пораженными генами наблюдаются дефекты генов, необходимых для функционирования внеклеточного матрикса . [ 9 ] Одной из таких форм является врожденная мышечная дистрофия с дефицитом мерозина (ВМД1А), на которую приходится около трети всех случаев ВМД и которая вызвана мутациями гена LAMA2 на хромосоме 6q2, кодирующего цепь ламинина-α2 . [ 10 ] [ 13 ] Ламинин-α2 является важной частью таких белков, как ламинин -2 и ламинин-4, которые выполняют важные функции в мышечных движениях, и у большинства пациентов с мутированным геном LAMA2 отсутствует экспрессия ламинина-α2 в мышечной ткани. [ 13 ] Другой формой в этой группе является врожденная мышечная дистрофия Ульриха , которая вызвана мутациями в генах COL6A1 , COL6A2 и COL6A3 , которые кодируют три альфа-цепи, составляющие коллаген VI . [ 11 ] [ 14 ] Коллаген VI важен для мышечной, сухожильной и кожной тканей и предназначен для прикрепления клеток к внеклеточному матриксу. [ 11 ] [ 14 ] ВМД Ульриха может быть вызвана как аутосомно-рецессивными, так и аутосомно-доминантными мутациями, хотя доминантные мутации обычно возникают de novo. [ 11 ] [ 14 ] Рецессивные мутации часто приводят к полному отсутствию коллагена VI во внеклеточном матриксе, тогда как существуют различные типы доминантных мутаций, которые могут вызывать частичную функцию коллагена V1. [ 11 ] [ 14 ]
Другой формой ВМД является врожденная мышечная дистрофия ригидного позвоночника (RSMD1), или синдром ригидного позвоночника , который вызван мутациями в гене SELENON, кодирующем селенопротеин N. [ 13 ] Точная функция селенопротеина N неизвестна, но он экспрессируется в шероховатой эндоплазматической сети скелетных мышц , сердца, головного мозга, легких и тканях плаценты, а также на высоких уровнях в диафрагме. [ 13 ] RSMD1 характеризуется осевой и дыхательной слабостью, ригидностью позвоночника и сколиозом , а также мышечной атрофией , и хотя это редкая форма CMD, мутации SEPN1 наблюдаются и при других врожденных миопатиях . [ 9 ]
Некоторыми из наиболее распространенных форм CMD являются дистрогликанопатии, вызванные дефектами гликозилирования α- дистрогликана (α-DG), который помогает связать внеклеточный матрикс и цитоскелет . [ 12 ] [ 15 ] Дистрогликанопатии вызваны мутациями в генах, кодирующих белки, участвующие в модификации α-DG после трансляции белка, а не мутациями в самом белке. [ 9 ] Было обнаружено 19 генов, вызывающих дистрофии, связанные с α-ДГ, с наблюдаемым широким спектром фенотипических эффектов, характеризующихся пороками развития головного мозга наряду с мышечной дистрофией. [ 12 ] [ 13 ] [ 15 ] Синдром Уокера-Варбурга (WWS) является наиболее тяжелым фенотипом дистрогликанопатии, при этом ген POMT1 является первым геном, вызывающим синдром Уокера-Варбурга (WWS), хотя существует 11 дополнительных генов, участвующих в WWS. Эти гены включают POMT2 , FKRP , FKTN , ISPD, CTDC2, TMEM5, POMGnT1 , B3GALnT2 , GMPPB , B3GnT1 и SGK196, многие из которых были идентифицированы как участвующие в других дистрогликанопатиях. [ 12 ] [ 15 ] У пациентов наблюдается мышечная слабость, пороки развития мозжечка и глаз, при этом ожидаемая продолжительность жизни составляет менее 1 года. [ 9 ] [ 15 ]
Дополнительным фенотипом дистрогликанопатии является врожденная мышечная дистрофия Фукуямы (ВМД), вызванная мутацией гена Фукутина (FKTN), которая является вторым наиболее распространенным типом мышечной дистрофии в Японии после мышечной дистрофии Дюшенна . [ 12 ] Мутацией-основателем FCMD является вставка ретротранспозона длиной 3 тыс . оснований в некодирующую область FKTN, что приводит к мышечной слабости, нарушению функции глаз, судорогам и умственной отсталости. [ 14 ] Хотя точная функция FKTN неизвестна, мРНК FKTN экспрессируется у плода в развивающейся ЦНС , мышцах и глазах и, вероятно, необходима для нормального развития, поскольку полная инактивация приводит к гибели эмбриона на 7-й день. [ 13 ] Другой фенотип, болезнь «мышцы-глаз-мозг» (MEB), является дистрогликанопатией, наиболее распространенной в Финляндии и вызван мутациями в генах POMGnT1, FKRP, FKTN, ISPD и TMEM5. [ 15 ] Ген POMGnT1 экспрессируется в тех же тканях, что и FKTN, а степень тяжести MEB, по-видимому, аналогична FCMD. [ 12 ] [ 13 ] Однако симптомы, уникальные для MEB, включают глаукому , атрофию зрительных нервов и образование сетчатки. [ 9 ] Наименее тяжелым фенотипом дистрогликанопатий является CMD типа 1c (MDC1C), обусловленный мутациями в гене FKRP и LARGE , с фенотипом, сходным с MEB и WWS. [ 15 ] MDC1C также включает мышечную дистрофию конечностей и пояса . [ 12 ] [ 15 ]
Механизм
[ редактировать ]Что касается механизма врожденной мышечной дистрофии, можно обнаружить, что, хотя существует много типов ВМД, гликозилирование и α-дистрогликана изменения в тех генах, которые вовлечены, являются важной частью патофизиологии этого состояния. [ 16 ]
Диагностика
[ редактировать ]Скелетно-мышечное обследование при врожденных мышечных дистрофиях.
[ редактировать ]Мышечный фиброз и контрактуры суставов или фиксированные деформации являются кардинальными клиническими признаками врожденных мышечных дистрофий. Фиброз и укорочение мышц в конечном итоге приводят к контрактурам суставов или фиксированным деформациям. Они важны для диагностики ВМД. Однако у некоторых пациентов изначально наблюдается слабость суставов. Деформации суставов могут возникать в конечностях и позвоночнике. Тяжелые деформации могут привести к вывихам суставов и затруднениям при ходьбе или нарушениям походки. [ 6 ] Однако конкретная картина поражения мышц при каждом из подтипов ВМД до конца не выяснена. Недавний обзор выявил специфичные для подтипа ВМД клинические закономерности поражения мышц и суставов, которые могут помочь в дифференциальной диагностике подтипов ВМД. Это особенно справедливо для врожденной мышечной дистрофии с дефицитом мерозина (MDC1A) или подтипа CMD, связанного с LAMA2. [ 6 ] Тем не менее, эти паттерны поражения мышц и суставов должны коррелировать с другими клиническими признаками, отчетами нейровизуализации, иммунным окрашиванием мышечной биопсии и результатами молекулярного или генетического анализа, если они доступны. Такой комплексный подход имеет решающее значение для правильной и своевременной диагностики ВМД. [ 6 ]
Кардиологическое исследование при врожденных мышечных дистрофиях
[ редактировать ]Кардиологические проявления ВМД сильно различаются. Они могут варьироваться от несуществующих или легких до тяжелых и фатальных поражений сердца. Как правило, сердечные нарушения при ВМД могут проявляться в виде дилатационной кардиомиопатии, систолической дисфункции, гипертрофической кардиомиопатии, фиброза миокарда или фатальных желудочковых аритмий. [ 17 ] [ 18 ] Дистрогликанопатии, такие как врожденная мышечная дистрофия Фукуямы, имеют относительно высокую вероятность развития значительных сердечных проявлений. При врожденной мышечной дистрофии с дефицитом мерозина (MDC1A) или ВМД, связанной с LAMA2, сердечные проявления обычно протекают бессимптомно. Сердечные проявления также были связаны с мышечной дистрофией конечностей 2I и ВМН, связанной с LMNA. Сердечные проявления могут быть вторичными по отношению к тяжелой деформации грудного отдела позвоночника, например, при синдроме ригидного позвоночника . [ 17 ] [ 18 ] Кардиологическое обследование и скрининг важны для пациентов с ВМД. Наблюдение важно для тех, у кого диагностировано поражение сердца.

Для диагностики врожденной мышечной дистрофии проводятся следующие тесты/обследования: [ 2 ]
- Лабораторное исследование ( уровни КК )
- мышц МРТ и особенно МРТ мышц всего тела недавно использовалась для описания мышечных аномалий у пациентов с первичным подтипом дефицита ламинина-α2 (мерозина) при ВМД.
- ЭМГ
- Генетическое тестирование
Классификация (различные виды врожденных мышечных дистрофий)
[ редактировать ]Подтипы врожденной мышечной дистрофии были установлены посредством вариаций в нескольких генах. фенотипы , а также классификации генотипов . В некоторой литературе для установления подтипов используются [ 1 ]
Установлено, что врожденные мышечные дистрофии могут быть как аутосомно-доминантными , так и аутосомно-рецессивными по типу наследования, хотя последний вариант встречается гораздо чаще. [ 1 ]
Лица с врожденной мышечной дистрофией относятся к одному из следующих типов :
- ВМД с мозго-глазным заболеванием , также называемым заболеванием «мышцы-глаз-мозг» , [ 19 ] Это редкая форма врожденной мышечной дистрофии (аутосомно-рецессивное заболевание), вызывающая отсутствие нормального мышечного тонуса, что может задерживать ходьбу из-за слабости, а также паралич глазных мышц и умственную отсталость, что влияет на способ обработки информации человеком. [ 19 ] Это вызвано мутацией гена POMGNT1 . [ 19 ]
- ВМД с отведенными (втянутыми внутрь) большими пальцами . редкая форма ВМД, вызывающая необратимое укорочение суставов пальцев ног и отсутствие мышечного тонуса, что может задерживать ходьбу из-за слабости человека. У человека с этой формой врожденной мышечной дистрофии в некоторых случаях может наблюдаться легкая гипоплазия мозжечка . [ 1 ]
- ВМД/ЛГМД без МР – первые годы жизни новорожденного начинаются со слабости, которая влияет на двигательные навыки, в подростковом возрасте может осуществляться ходьба, деформация и ригидность суставов. Суставы, шея и позвоночник; прогрессирующая кардиомиопатия в раннем возрасте; У человека могут присутствовать нарушения сердечного ритма. [ 1 ]
- Большие связанные с CMD в начале периода новорожденности проблемы, которые получает ребенок; плохой мышечный тонус и слабая двигательная функция; у человека появится умственная отсталость , а структура мозга, скорее всего, будет ненормальной. [ 20 ]
- ВМД с атрофией мозжечка , тяжелая гипоплазия мозжечка, плохой мышечный тонус, задержка двигательных функций, отсутствие координации двигательных навыков, трудности с речью, непроизвольные движения и некоторая умственная отсталость. Кроме того, мышечная биопсия не выявляет каких-либо нарушений. [ 1 ]
- Синдром Уокера-Варбурга вначале прогрессирующая слабость и низкий мышечный тонус при рождении или в раннем детстве; мелкие мышцы; большинство заболевших детей не доживают до 3-летнего возраста. Имеются проблемы со структурой глаз, сопровождающиеся ухудшением зрения. [ 21 ]
- При ВМД с первичным дефицитом ламинина-α2 (мерозина) (MDC1A) интеллект у таких лиц не нарушен, наблюдается ослабление проксимальных мышц и ригидность позвоночника, а также поражение дыхательных путей (при прогрессировании заболевания). [ 22 ]
- CMD/LGMD с МР- слабостью, деформацией и ригидностью суставов, присутствующими при рождении, плохим мышечным тонусом, медленно прогрессирующим; у людей могут наблюдаться кисты мозжечка (или кортикальные проблемы), микроцефалия также может присутствовать . Может возникнуть ненормальная гибкость, возможно искривление позвоночника. [ 1 ]
- CDG I (DPM3): некоторыми симптомами при рождении и на протяжении всей жизни ребенка являются слабость или плохой мышечный тонус. повышение уровня креатинкиназы У человека может наблюдаться кардиомиопатия (отсутствие обструкции оттока крови), также может наблюдаться в сыворотке. Могут присутствовать некоторые проблемы с IQ, а также слабость проксимальных мышц. Также следует отметить снижение содержания долихолфосфата маннозы . [ 23 ]
- CDG I (DPM2) слабый мышечный тонус, начиная с первых недель жизни ребенка, у человека могут проявляться тяжелые неврологические физические характеристики, которые приводят к летальному исходу в раннем возрасте. гипотония У таких лиц могут присутствовать и миопатическое лицо, а также контрактуры суставов. Наконец, миоклонические судороги могут возникать в очень раннем возрасте (3 месяца). [ 24 ]
- CDG Ie (DPM1) при рождении у ребенка будет слабость с поражением дыхательной системы, а также серьезные психические и психомоторные проблемы. К 3 годам человек может стать слепым и иметь проблемы с речью. Микроцефалия может возникнуть в раннем детстве, как и судороги. [ 25 ]
- CMD с ригидностью позвоночника, присутствующей при рождении, может иметь плохой мышечный тонус и слабость, снижение дыхательной способности, мышцы могут быть деформированы, начиная с раннего возраста, стабилизируется или медленно снижается ригидность позвоночника, ограниченная подвижность при сгибании шеи и позвоночника, искривление позвоночника и прогрессирующая деформация и ригидность. суставов, незначительные нарушения сердечной деятельности , нормальный интеллект. [ 26 ]

- CMD с аномалией ламина A/C : в первый год ребенок слаб, позже у человека могут возникнуть проблемы с поднятием рук и головы. Может потребоваться назогастральный зонд , слабость конечностей и повышение уровня креатинкиназы в сыворотке. Человек может демонстрировать диафрагмальный характер дыхания. [ 27 ]
- Слабость интегрина α7 , присутствующая при рождении, плохой мышечный тонус при поздней ходьбе, потеря мышечной ткани, умственная отсталость. Кроме того, уровень креатинкиназы был повышен. [ 28 ]
- Фукуяма КМД – в западных странах этот тип КМД встречается редко, но распространен в Японии. Последствия этого заболевания для младенцев варьируются по степени тяжести. К ним относятся слабость мышечного тонуса в течение первого года, деформированные и ригидные суставы, искривления позвоночника, судороги, поражение глаз и умственная отсталость. Некоторые пациенты могут достичь ограниченной подвижности при ходьбе. [ 29 ]
- Мерозин-дефицитная ВМД – слабость мышечного тонуса, присутствующая при рождении, спектр тяжести; может проявляться гипотония и плохое моторное развитие. У большинства людей наблюдаются перивентрикулярные проблемы с белым веществом. Однако в большинстве случаев умственная отсталость встречается редко. [ 30 ]
- Мерозин-положительная ВМД. Некоторыми формами мерозин-положительной ВМД являются: ранняя ригидность позвоночника, ВМД с мышечной гипертрофией, ВМД с мышечной гипертрофией и дыхательной недостаточностью. [ 31 ]

- Врожденная мышечная дистрофия Ульриха при рождении проявляется слабостью и плохим мышечным тонусом.
Дифференциальный диагноз
[ редактировать ]DDx врожденной мышечной дистрофии у больного человека выглядит следующим образом (существуют также ненервно-мышечные генетические состояния). [ 33 ] ): [ 2 ]
Управление
[ редактировать ]
Что касается лечения врожденной мышечной дистрофии, Американская академия неврологии рекомендует людям Необходимо проводить мониторинг сердечной функции, дыхания и желудочно-кишечного тракта . Кроме того, считается, что терапия в речевой, ортопедической и физической областях улучшит качество жизни человека. [ 3 ]
Хотя в настоящее время лекарства не существует, важно сохранить мышечную активность и любую доступную коррекцию скелетных аномалий (например, сколиоза). Ортопедические процедуры, такие как спондилодез , поддерживают/увеличивают шансы человека на большее физическое движение. [ 3 ]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Перейти обратно: а б с д и ж г час я дж Спаркс, Сьюзен; Кихано-Рой, Сусана; Харпер, Эми; Рутковски, Энн; Гордон, Эринн; Хоффман, Эрик П.; Пегораро, Елена (1 января 1993 г.). «Обзор врожденной мышечной дистрофии – УДАЛЕНАЯ ГЛАВА, ТОЛЬКО ДЛЯ ИСТОРИЧЕСКОЙ СПРАВКИ». В Пагоне, Роберта А.; Адам, Маргарет П.; Ардингер, Холли Х.; Уоллес, Стефани Э.; Амемия, Энн; Бин, Лора Дж. Х.; Берд, Томас Д.; Фонг, Чин-То; Меффорд, Хизер С. (ред.). Обзор врожденной мышечной дистрофии . Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл. ПМИД 20301468 . Архивировано из оригинала 04 августа 2020 г. Проверено 30 августа 2017 г. обновление 2012
- ^ Перейти обратно: а б с «Обследование врожденной мышечной дистрофии: лабораторные исследования, визуализирующие исследования, другие тесты» . emedicine.medscape.com . Архивировано из оригинала 19 января 2021 г. Проверено 28 апреля 2016 г.
- ^ Перейти обратно: а б с «Врожденная мышечная дистрофия» . Рекомендации Американской академии неврологии . 2015. Архивировано из оригинала 19 октября 2020 года . Проверено 28 апреля 2016 г.
- ^ Бертини, Энрико; Д'Амико, Адель; Гуаланди, Франческа; Петрини, Стефания (01 декабря 2011 г.). «Врожденные мышечные дистрофии: краткий обзор» . Семинары по детской неврологии . 18 (4): 277–288. дои : 10.1016/j.spen.2011.10.010 . ISSN 1071-9091 . ПМЦ 3332154 . ПМИД 22172424 .
- ^ «Гипотония: Медицинская энциклопедия MedlinePlus» . www.nlm.nih.gov . Архивировано из оригинала 5 июля 2016 г. Проверено 28 апреля 2016 г.
- ^ Перейти обратно: а б с д Эль-Собкий, Тамер А.; Абдулхади, Хала; Махмуд, Шейди; Аминь, Джон (31 января 2024 г.). «Ортопедические проявления подтипов врожденной мышечной дистрофии у детей: новые признаки нуждаются в консолидации: обзорный обзор». Журнал скелетно-мышечной хирургии и исследований . 8 : 11–23. дои : 10.25259/JMSR_229_2023 .
- ^ Астрея, Гуджа; Баттини, Роберта; Лензи, Сара; Фросини, Сильвия; Бонетти, Сильвия; Моретти, Елена; Перацца, Сильвия; Санторелли, Филиппо М.; Печини, Кьяра (октябрь 2016 г.). «Нарушение обучаемости при нервно-мышечных расстройствах: трамплин во взрослую жизнь» . Акта Миологика . 35 (2): 90–95. ISSN 1128-2460 . ПМЦ 5343745 . ПМИД 28344438 .
- ^ Перейти обратно: а б Замбон, Альберто А.; Мунтони, Франческо (01 октября 2021 г.). «Врожденные мышечные дистрофии: что нового?» . Нервно-мышечные расстройства . 31 (10): 931–942. дои : 10.1016/j.nmd.2021.07.009 . ISSN 0960-8966 . ПМИД 34470717 . S2CID 236462260 .
- ^ Перейти обратно: а б с д и ж Киршнер, Янбернд (1 января 2013 г.), Дюлак, Оливье; Лассонд, Мариз; Сарнат, Харви Б. (ред.), «Глава 143 - Врожденные мышечные дистрофии» , Справочник по клинической неврологии , Детская неврология, часть III, 113 , Elsevier: 1377–1385, doi : 10.1016/b978-0-444-59565-2.00008 -3 , ISBN 9780444595652 , PMID 23622361 , заархивировано из оригинала 4 октября 2023 г. , получено 11 мая 2022 г.
- ^ Перейти обратно: а б Адам, член парламента; Мирзаа, генеральный менеджер; Пагон, РА; Уоллес, ЮВ; Бин, LJH; Грипп, К.В.; Амемия, А; Оливейра, Дж; Паренте Фрейшо, Дж; Сантос, М; Коэльо, Т. (1993). «Мышечная дистрофия LAMA2». ПМИД 22675738.
- ^ Перейти обратно: а б с д и Адам, член парламента; Мирзаа, генеральный менеджер; Пагон, РА; Уоллес, ЮВ; Бин, LJH; Грипп, К.В.; Амемия, А; Фоли, Арканзас; Моассель, П; Донкерворт, С; Болдук, В; Беннеманн, К.Г. (1993). «Дистрофии, связанные с коллагеном VI». ПМИД 20301676.
- ^ Перейти обратно: а б с д и ж г Сайто, Кайоко (1993). «Врожденная мышечная дистрофия Фукуямы» . GeneReviews® . Вашингтонский университет, Сиэтл. ПМИД 20301385 . Архивировано из оригинала 18 сентября 2019 г. Проверено 4 октября 2023 г.
- ^ Перейти обратно: а б с д и ж г час Хименес-Маллебрера, К.; Браун, Южная Каролина; Сьюри, Калифорния; Мунтони, Ф. (1 апреля 2005 г.). «Врожденная мышечная дистрофия: молекулярные и клеточные аспекты» . Клеточные и молекулярные науки о жизни . 62 (7): 809–823. дои : 10.1007/s00018-004-4510-4 . ISSN 1420-9071 . ПМИД 15868406 . S2CID 24662420 .
- ^ Перейти обратно: а б с д и Беннеманн, Карстен Г. (июль 2011 г.). «Миопатии, связанные с коллагеном VI: мышца встречается со своим матриксом» . Обзоры природы Неврология . 7 (7): 379–390. дои : 10.1038/nrneurol.2011.81 . ISSN 1759-4766 . ПМК 5210181 . ПМИД 21691338 .
- ^ Перейти обратно: а б с д и ж г Фу, Сяо-На; Сюн, Хуэй (05.11.2017). «Генетические и клинические достижения врожденной мышечной дистрофии» . Китайский медицинский журнал . 130 (21): 2624–2631. дои : 10.4103/0366-6999.217091 . ISSN 0366-6999 . ПМЦ 5678264 . ПМИД 29067961 .
- ^ Мартин, Пол Т. (2006). «Механизмы заболевания: врожденные мышечные дистрофии — гликозилирование занимает центральное место» . Природная клиническая практика Неврология . 2 (4): 222–230. дои : 10.1038/ncpneuro0155 . ISSN 1745-834X . ПМЦ 2855642 . ПМИД 16932553 .
- ^ Перейти обратно: а б Бэдилэ, Э; Лунгу, II; Грумесеску, AM; Скафа Удриште, А (12 мая 2021 г.). «Диагностика нарушений сердечной деятельности при мышечных дистрофиях» . Медицина (Каунас, Литва) . 57 (5): 488. doi : 10.3390/medicina57050488 . ПМК 8151418 . ПМИД 34066119 .
- ^ Перейти обратно: а б Финстерер, Дж; Рамачиотти, К; Ван, Швейцария; Вахби, К; Розенталь, Д; Дубок, Д; Мелачини, П. (сентябрь 2010 г.). «Кардиологические данные при врожденных мышечных дистрофиях». Педиатрия . 126 (3): 538–45. дои : 10.1542/пед.2010-0208 . ПМИД 20679303 .
- ^ Перейти обратно: а б с "Запись OMIM - #253280 - МЫШЕЧНАЯ ДИСТРОФИЯ-ДИСТРОГЛИКАНОПАТИЯ (ВРОЖДЕННАЯ С АНОМАЛИЯМИ МОЗГА И ГЛАЗ), ТИП А, 3; MDDGA3" . www.omim.org . Архивировано из оригинала 11 декабря 2015 г. Проверено 26 апреля 2016 г.
- ^ «Ошибка 403» .
- ^ Справочник, Дом генетики. «Синдром Уокера-Варбурга» . Домашний справочник по генетике . Архивировано из оригинала 28 июля 2018 г. Проверено 26 апреля 2016 г.
- ^ Кихано-Рой, Сусана; Спаркс, Сьюзен; Рутковски, Энн (1 января 1993 г.). «Мышечная дистрофия LAMA2». В Пагоне, Роберта А.; Адам, Маргарет П.; Ардингер, Холли Х.; Уоллес, Стефани Э.; Амемия, Энн; Бин, Лора Дж. Х.; Берд, Томас Д.; Фонг, Чин-То; Меффорд, Хизер С. (ред.). Мышечная дистрофия, связанная с LAMA2 . Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл. ПМИД 22675738 . обновление 2012
- ^ «Запись OMIM - # 612937 - ВРОЖДЕННОЕ НАРУШЕНИЕ ГЛИКОЗИЛИРОВАНИЯ, ТИП Io; CDG1O» . www.omim.org . Архивировано из оригинала 8 июня 2019 г. Проверено 26 апреля 2016 г.
- ^ «Запись OMIM - # 615042 - ВРОЖДЕННОЕ НАРУШЕНИЕ ГЛИКОЗИЛИРОВАНИЯ, ТИП Iu; CDG1U» . www.omim.org . Архивировано из оригинала 23 июня 2019 г. Проверено 26 апреля 2016 г.
- ^ «Запись OMIM - # 608799 - ВРОЖДЕННОЕ НАРУШЕНИЕ ГЛИКОЗИЛИРОВАНИЯ, ТИП Ie; CDG1E» . www.omim.org . Архивировано из оригинала 29 сентября 2019 г. Проверено 26 апреля 2016 г.
- ^ «Запись OMIM - № 602771 - ЖЕСТКАЯ МЫШЕЧНАЯ ДИСТРОФИЯ ПОЗВОНОЧНИКА 1; RSMD1» . www.omim.org . Архивировано из оригинала 7 декабря 2015 г. Проверено 26 апреля 2016 г.
- ^ «Запись OMIM — № 613205 — МЫШЕЧНАЯ ДИСТРОФИЯ ВРОЖДЕННАЯ, СВЯЗАННАЯ с LMNA» . www.omim.org . Архивировано из оригинала 18 сентября 2019 г. Проверено 26 апреля 2016 г.
- ^ «Запись OMIM - # 613204 - МЫШЕЧНАЯ ДИСТРОФИЯ ВРОЖДЕННАЯ, ВСЛЕДСТВИЕ ДЕФИЦИТА ИНТЕГРИНА АЛЬФА-7» . www.omim.org . Архивировано из оригинала 15 декабря 2021 г. Проверено 26 апреля 2016 г.
- ^ Справочник, Дом генетики. «Врожденная мышечная дистрофия Фукуямы» . Домашний справочник по генетике . Архивировано из оригинала 13 мая 2016 г. Проверено 26 апреля 2016 г.
- ^ «Запись OMIM - # 607855 - МЫШЕЧНАЯ ДИСТРОФИЯ, ВРОЖДЕННЫЙ ДЕФИЦИТ МЕРОЗИНА, 1A; MDC1A» . www.omim.org . Архивировано из оригинала 14 апреля 2020 г. Проверено 26 апреля 2016 г.
- ^ «Запись OMIM - % 609456 - МЫШЕЧНАЯ ДИСТРОФИЯ ВРОЖДЕННАЯ, МЕРОЗИН-ПОЛОЖИТЕЛЬНАЯ» . www.omim.org . Архивировано из оригинала 15 декабря 2021 г. Проверено 26 апреля 2016 г.
- ^ «Запись OMIM — № 254090 — ВРОЖДЕННАЯ МЫШЕЧНАЯ ДИСТРОФИЯ УЛЛРИХА 1; UCMD1» . omim.org . Архивировано из оригинала 24 декабря 2015 г. Проверено 26 апреля 2016 г.
- ^ Боннеманн, Карстен Г.; Ван, Чинг Х.; Кихано-Рой, Сьюзен; Деконинк, Николас; Бертини, Генри; Феррейро, Ханна; Мантон, Франческо; Сьюри, Кэролайн; Беруд, Кристофер; Мэтьюз, Кэтрин Д.; Мур, Стивен А.; Беллини, Джонатан; Рутковски, Энн; Норт, Кэтрин Н. (1 апреля 2014 г.). «Диагностический подход к врожденным мышечным дистрофиям» . Нервно-мышечные расстройства . 24 (4): 289–311. дои : 10.1016/j.nmd.2013.12.011 . ПМК 5258110 . ПМИД 24581957 .
Дальнейшее чтение
[ редактировать ]- А, Грациано; Ф, Бьянко; А, Д'Амико; я, Мороний; С, Мессина; С, Бруно; Э, Пегораро; М, Мора; Г, Астрея (01 марта 2015 г.). «Распространенность врожденной мышечной дистрофии в Италии: популяционное исследование» . Неврология . 84 (9): 904–911. дои : 10.1212/WNL.0000000000001303 . ISSN 0028-3878 . ПМЦ 4351663 . ПМИД 25653289 .
- Пако, Соня; Кассеррас, Тереза; Родригес, Мария Анхельс; Джоу, Кристина; Пучделлос, Монтсеррат; Ортес, Карлос И.; Диас-Манера, Хорди; Галлардо, Эдуардо; Коломер, Жауме (15 декабря 2015 г.). «Транскриптомный анализ фибробластов врожденной мышечной дистрофии Ульриха выявляет сигнатуру внеклеточного матрикса заболевания и ключевые молекулярные регуляторы» . ПЛОС ОДИН . 10 (12): e0145107. Бибкод : 2015PLoSO..1045107P . дои : 10.1371/journal.pone.0145107 . ISSN 1932-6203 . ПМЦ 4686057 . ПМИД 26670220 .
- Фальсаперла, Рафаэле; Пратико, Андреа Д.; Руджери, Мартино; Парано, Энрико; Риццо, Рената; Корселло, Джованни; Виталити, Джованна; Павоне, Пьеро (31 августа 2016 г.). «Врожденная мышечная дистрофия: от мышц к мозгу» . Итальянский журнал педиатрии . 42 (1): 78. дои : 10.1186/s13052-016-0289-9 . ISSN 1824-7288 . ПМК 5006267 . ПМИД 27576556 .
- «Краткое содержание научно обоснованного руководства для ПАЦИЕНТОВ И ИХ СЕМЕЙ С ВРОЖДЕННОЙ МЫШЕЧНОЙ ДИСТРОФИЯЮ» . ааан.com . Американская академия неврологии (ААН) . Проверено 5 декабря 2017 г.