Недержание пигмента
| Недержание пигмента | |
|---|---|
| Другие имена | Синдром Блоха-Сименса, болезнь Блоха-Сульцбергера, синдром Блоха-Сульцбергера, неланобластоз кожи, систематический пигментный невус [1] |
 | |
| Это состояние наследуется по Х-сцепленному доминантному типу. | |
| Специальность | Медицинская генетика |
Пигментное недержание ( ИП ) — редкое Х-сцепленное доминантное генетическое заболевание , поражающее кожу, волосы, зубы, ногти и центральную нервную систему. Он получил свое название от внешнего вида под микроскопом. [1]
Заболевание характеризуется кожными аномалиями, которые начинаются в детстве, обычно в виде волдырей, которые заживают, с последующим развитием более твердых кожных новообразований. На коже могут появиться серые или коричневые пятна, которые со временем бледнеют. Другие симптомы могут включать выпадение волос, аномалии зубов, аномалии глаз, которые могут привести к потере зрения, а также покрытые морщинами или ямками ногти на руках и ногах. Сопутствующие проблемы могут включать задержку развития, умственную отсталость, судороги и другие неврологические проблемы. Большинство мужчин с этим заболеванием не доживают до родов.
Пигментное недержание вызвано мутацией гена IKBKG , который кодирует белок NEMO, который служит для защиты клеток от TNF-альфа индуцированного апоптоза, . Таким образом, недостаток IKBKG делает клетки более склонными к апоптозу.
Специфического лечения не существует; индивидуальные условия должны управляться специалистами. [2]
Презентация
[ редактировать ]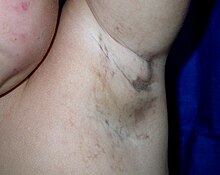
Поражения кожи развиваются характерными стадиями: [ нужна ссылка ]
- образование волдырей (от рождения до четырехмесячного возраста),
- сыпь бородавчатая (в течение нескольких месяцев),
- вихревая макулярная гиперпигментация (приблизительно с шестимесячного возраста и во взрослом возрасте), за которой следует
- линейная гипопигментация .
алопеция Наблюдаются , зубные аномалии, дистрофия ногтей. У некоторых пациентов наблюдаются сосудистые нарушения сетчатки, предрасполагающие к отслойке сетчатки в раннем детстве. когнитивные задержки или умственная отсталость . Иногда наблюдаются [ нужна ссылка ]
Обесцвеченная кожа вызвана чрезмерным отложением меланина (нормального пигмента кожи ).У большинства новорожденных с IP в течение первых двух недель меняется цвет кожи.Пигментация затрагивает туловище и конечности , она грифельно-серая, синяя или коричневая, распределена неравномерными мраморными или волнистыми линиями.Изменение цвета иногда исчезает с возрастом. [ нужна ссылка ]
Неврологические проблемы могут включать церебральную атрофию , образование небольших полостей в центральном белом веществе головного мозга и потерю нейронов в коре мозжечка .Около 20% детей с ИП будут иметь замедленное моторное развитие , мышечную слабость на одной или обеих сторонах тела, умственную отсталость и судороги.У них также могут быть проблемы со зрением, которые могут включать в себя: косоглазие , катаракту , отслойку сетчатки и тяжелую потерю зрения.Проблемы с зубами также распространены и могут включать гиподентию , зубы неправильной формы и задержку прорезывания зубов . [3]
Аномалии молочной железы могут встречаться у 1% пациенток и включать гипоплазию или избыточные соски .
Скелетные и структурные аномалии могут возникать примерно у 14% пациентов, в том числе: [ нужна ссылка ]
- Соматическая асимметрия
- полупозвонки
- Сколиоз
- Расщелина позвоночника
- Синдактилия
- Ахейрия ( врожденное отсутствие рук – примечание: могут поражаться и другие конечности)
- Аномалии уха
- Дополнительные ребрышки
- Деформации черепа
Генетика
[ редактировать ]IP наследуется по Х-сцепленному доминантному типу. [4] [5] IP смертелен для большинства, но не для всех мужчин. Женщина с IP может унаследовать мутацию IKBKG от любого из родителей или иметь новую генную мутацию. Родители могут быть либо клинически поражены, либо иметь зародышевый мозаицизм . Затронутые женщины имеют 50% риск передачи мутантного аллеля IKBKG при зачатии; однако у большинства зачатых мужчин происходит выкидыш. Таким образом, эффективное соотношение живорожденных детей от матери-носителя мутации составляет 33% незатронутых женщин, 33% пораженных женщин и 33% незатронутых мальчиков. генетическое консультирование , пренатальное тестирование и преимплантационная генетическая диагностика . Доступны [ нужна ссылка ]
У женщин клетки, экспрессирующие мутированный ген IKBKG из-за лионизации, избирательно умирают примерно во время рождения, поэтому Х-инактивация крайне искажена . [6]
ИП вызван мутациями в гене NEMO ( незаменимый модулятор NF-κB ). [ нужна ссылка ]
Диагностика
[ редактировать ]Диагноз ИП устанавливается на основании клинических данных и иногда с помощью подтверждающей биопсии кожи. Молекулярно -генетическое тестирование гена NEMO IKBKG (хромосомный локус Xq28) выявляет мутации, вызывающие заболевание, примерно у 80% пробандов. Такое тестирование доступно клинически. Кроме того, у женщин с ИП наблюдается искаженная инактивация Х-хромосомы; тестирование на это может быть использовано для подтверждения диагноза. Многим людям в прошлом ошибочно диагностировали второй тип IP, ранее известный как IP1. Сейчас этому заболеванию присвоено собственное название: гипомеланоз Ито ( incontentia пигментные ахромии ). Это имеет несколько иное представление: завитки или полосы гипопигментации и депигментации. Он не передается по наследству и не поражает кожу 1 или 2 стадии. Около 33–50% пациентов имеют мультисистемное поражение — глазные, скелетные и неврологические нарушения. Его хромосомный локус находится в Xp11, а не в Xq28. [ нужна ссылка ]
Уход
[ редактировать ]Специального лечения ИП пока не существует. Лечение может устранять только отдельные симптомы. [7]
История
[ редактировать ]Об этом заболевании впервые сообщили швейцарский дерматолог Бруно Блох в 1926 году и американский дерматолог Марион Сульцбергер в 1928 году. [8] [9] [2]
См. также
[ редактировать ]- Список кожных заболеваний
- Список рентгенографических данных, связанных с кожными заболеваниями
- Список стоматологических аномалий, связанных с кожными заболеваниями
Ссылки
[ редактировать ]- ^ Jump up to: а б Рапини, Рональд П.; Болонья, Жан Л.; Хориццо, Джозеф Л. (2007). Дерматология: Набор из 2 томов . Сент-Луис: Мосби. ISBN 978-1-4160-2999-1 . [ нужна страница ]
- ^ Jump up to: а б Сульцбергер, Мэрион Б. (1928). «О ранее описанной врожденной пигментной аномалии». Архив дерматологии и сифилиса (на немецком языке). 154 : 19–32. дои : 10.1007/bf01828398 . S2CID 40446256 .
- ^ Минич, С; Трпинац, Д; Габриэль, Х; Генчик, М; Обрадович, М. (январь 2013 г.). «Дентальные и оральные аномалии при пигментном недержании: систематический обзор». Клин Оральное расследование . 17 (1): 1–8. дои : 10.1007/s00784-012-0721-5 . ПМИД 22453515 . S2CID 73197872 .
- ^ Петтигрю, Рэйчел; Куо, Хун-Чжи; Скривен, Пол; Роуэлл, Паула; Пал, Кальяни; Хэндисайд, Алан; Брауде, Питер; Огилви, Кэролайн Маки (2000). «Беременность после ПГД по поводу Х-сцепленного аутосомно-доминантного недержания пигмента (синдром Блоха-Сульцбергера): отчет о случае» . Репродукция человека . 15 (12): 2650–2. дои : 10.1093/humrep/15.12.2650 . ПМИД 11098039 .
- ^ «Пигмент при недержании. DermNet NZ» .
- ^ Международный консорциум пигментной недержания (IP); Смахи, Асмаэ; Куртуа, Ж; Вабрес, П; Ямаока, С; Хойерц, С; Миних, А; Израиль, А; Хейсс, Нина С; Клаук, С.М.; Киошис, П; Виманн, С; Пустка, А; Эспозито, Тереза; Бардаро, Т; Джанфранческо, Ф; Чиккодикола, А; д'Урсо, М; Воффендин, Хейли; Джейкинс, Т; Доннаи, Д; Стюарт, Х; Кенрик, С.Дж.; Арадхья, Сваруп; Ямагата, Т; Леви, М; Льюис, Р.А.; Нельсон, Д.Л. (2000). «Геномная перестройка у NEMO нарушает активацию NF-κB и является причиной пигментного недержания» . Природа . 405 (6785): 466–72. Бибкод : 2000Natur.405..466T . дои : 10.1038/35013114 . ПМИД 10839543 . S2CID 186243924 .
- ^ «Недержание пигмента» . Медлайн Плюс Проверено 26 декабря 2017 г.
- ^ Пигментный дерматоз Блоха-Сульцбергера (Бруно Блох) в Who Named It?
- ^ Блох, Б. (1926). «Свойственное, до сих пор необъяснимое поражение пигмента (inконтинентия пигмента)». Швейцарский медицинский еженедельник (на немецком языке). 56 . Базель: 404–5.