синдром Ваарденбурга
| синдром Ваарденбурга | |
|---|---|
| Другие имена | Синдром Клейна-Ваарденбурга (тип 3), синдром Шаха-Ваарденбурга (тип 4) |
 | |
| Черты лица при синдроме Ваарденбурга 1 типа (из Яна ван дер Хуве , 1916 г.) описания | |
| Специальность | Медицинская генетика |
Синдром Ваарденбурга — это группа редких генетических состояний, характеризующихся по крайней мере некоторой степенью врожденной потери слуха и нарушений пигментации, которые могут включать ярко-голубые глаза (или один голубой и один карий глаз ), белый чуб или участки светлой кожи. Эти основные признаки составляют тип 2 состояния; при типе 1 имеется также более широкий промежуток между внутренними углами глаз , называемый телекантусом или дистопией канторум . [1] При типе 3, который встречается редко, руки также имеют деформацию, с постоянными контрактурами пальцев или сросшимися пальцами , а при типе 4 у человека также наблюдается болезнь Гиршпрунга . [2] [3] Также существует как минимум два типа (2E и PCWH), которые могут приводить к таким симптомам со стороны центральной нервной системы (ЦНС), как задержка развития и нарушения мышечного тонуса . [4]
Синдром вызван мутациями любого из нескольких генов, которые влияют на деление и миграцию клеток нервного гребня во время эмбрионального развития (хотя некоторые из вовлеченных генов также влияют на нервную трубку ). [5] Клетки нервного гребня — это стволовые клетки, оставшиеся после закрытия нервной трубки, которые формируют разнообразные клетки, не относящиеся к ЦНС, в различных частях тела, включая меланоциты , различные кости и хрящи лица, внутреннего уха и периферические нервы. кишечник . [6] Тип 1 вызван мутацией гена PAX3 , тогда как ген, который чаще всего вызывает тип 2 при мутации, — это MITF . [1] [7] Тип 3 является более тяжелым проявлением типа 1 и вызван мутацией того же гена, тогда как тип 4 чаще всего вызывается мутацией SOX10 . [2] [8] Мутации в других генах также могут вызывать различные типы, и некоторым из них были присвоены собственные буквенные подтипы. Большинство типов являются аутосомно-доминантными .
По оценкам, распространенность синдрома Ваарденбурга составляет 1 на 42 000. [5] [8] Типы 1 и 2 являются наиболее распространенными и составляют примерно половину и треть случаев соответственно, тогда как тип 4 составляет пятую часть, а тип 3 — менее 2% случаев. [8] По оценкам, 2–5% врожденно глухих людей страдают синдромом Ваарденбурга. [8] Описания синдрома датируются как минимум первой половиной 20 века, однако он назван в честь голландского офтальмолога и генетика Петруса Йоханнеса Ваарденбурга , описавшего его в 1951 году. [9] [10] Его подтипы постепенно открывались в последующие десятилетия, а гены, связанные с ними, приписывались в основном в 1990-х и 2000-х годах.
Признаки и симптомы
[ редактировать ]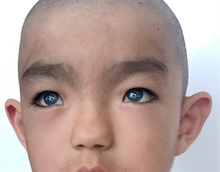
Синдром Ваарденбурга имеет несколько различных типов с некоторыми различиями в симптомах, причем симптомы могут различаться у людей одного и того же типа. Двумя особенностями, характерными для всех типов синдрома Ваарденбурга, являются некоторая степень врожденной нейросенсорной тугоухости и некоторая степень нарушений пигментации, наиболее часто встречающихся в глазах. [11]
Тип 1
[ редактировать ]Тип 1 характеризуется врожденной сенсоневральной тугоухостью , пигментными недостатками волос, такими как белая прядь волос ( полиоз ) в передней части головы или преждевременное поседение, пигментными недостатками глаз, такими как разноцветные глаза (полное гетерохромия радужной оболочки ), множественный цвет глаз (секторальная гетерохромия радужной оболочки) или ярко-голубые глаза, участки депигментации кожи и более широкий промежуток между внутренними углами глаз, называемый телекантусом или дистопией канторум. Другие черты лица, связанные с типом 1, могут включать высокую переносицу, плоский кончик носа, монобровь (синофрис), меньшие края ноздрей (крылья) или гладкий желобок . [1]
Тип 2
[ редактировать ]
Отличие типа 2 от типа 1 состоит в том, что у пациентов нет более широкой щели между внутренними углами глаз (телекантус/дистопия канторум). Нейросенсорная тугоухость этого типа встречается чаще и является более тяжелой. [7] [12] На сегодняшний день наиболее распространенным геном, вызывающим этот тип мутаций, является MITF (классифицируется как тип 2А). Если у двух лиц с мутацией в этом гене ( гетерозиготный ) есть ребенок, несущий обе мутации ( гомозиготный ), вероятность возникновения которого составляет 25%, у ребенка присутствуют дополнительные симптомы, такие как отверстие в радужной оболочке ( колобома ), маленькие глаза ( микрофтальмия ), затвердевшие кости ( остепетроз ), макроцефалия , альбинизм и глухота. [7]
Были известны два пациента с мутациями в обеих копиях SNAI2 (классифицируемых как тип 2D); у этих людей был синдром Ваарденбурга 2-го типа, но не было нарушений пигментации волос. [13]
Когда синдром Ваарденбурга 2-го типа вызван мутацией SOX10 (классифицируется как тип 2E), в некоторых случаях он может проявляться множественными неврологическими симптомами. К ним могут относиться задержка развития, нистагм в раннем детстве , повышенный мышечный тонус , белого вещества аномалии или гипомиелинизация головного мозга, аутистическое поведение и недоразвитие или полное отсутствие многих структур внутреннего уха, таких как вестибулярная система или улитка . Также может присутствовать отсутствие обоняния ( аносмия ) из-за отсутствия обонятельной луковицы в мозгу. [4]
Тип 3
[ редактировать ]Также известный как синдром Клейна-Ваарденбурга или синдром Ваарденбурга-Клейна, тип 3 имеет те же симптомы, что и тип 1 (и вызван мутациями в том же гене), но имеет дополнительные симптомы, которые поражают руки и кисти. К ним могут относиться контрактуры суставов пальцев ( камптодактилия ), обусловленные недоразвитием мышц, а также сросшиеся пальцы ( синдактилия ) или крылатые лопатки . Возможны также микроцефалия и задержка развития. [2]
Тип 4
[ редактировать ]Также известный как синдром Шаха-Ваарденбурга или синдром Ваарденбурга-Шаха, тип 4 имеет большинство тех же особенностей, что и тип 2 (т.е. нет телекантуса или явно более широкой глазной щели), но с добавлением болезни Гиршпрунга , которая является врожденным недостатком. нервов в кишечнике, что приводит к дисфункции кишечника . [3] [14] Кроме того, потеря слуха встречается не так часто, как при типе 2. [3] Редко заячьей губе при этой форме синдрома Ваарденбурга. сообщалось о [15]
Тип 4 также может быть вызван мутацией SOX10 (тот же ген, что и у типа 2E), в котором он известен как тип 4C; Потеря слуха этого типа очень распространена и серьезна. [16]
ПКВХ
[ редактировать ]Мутация SOX10 , гена, участвующего в типах 2E и 4C, иногда может приводить к появлению симптомов обоих типов (неврологические симптомы, иногда наблюдаемые при типе 2E, и болезнь Гиршпрунга, наблюдаемая при типе 4). Когда это происходит, это называется периферическая демиелинизирующая нейропатия – центральная дисмиелинизирующая лейкодистрофия – синдром Ваарденбурга – болезнь Гиршпрунга (ПКВГ). [17] [18]
Причина
[ редактировать ]
Синдром Ваарденбурга вызван мутациями в любом из нескольких генов, которые влияют на работу клеток нервного гребня в эмбриональном развитии . Большинство типов синдрома Ваарденбурга вызваны аутосомно-доминантными мутациями. Те немногие, которые являются аутосомно-рецессивными, встречаются редко. В большинстве случаев больной унаследовал заболевание от одного из родителей с одной из доминирующих форм заболевания. Небольшой процент случаев возникает в результате спонтанных новых мутаций в гене, при этом семейный анамнез заболевания отсутствует. [ нужна ссылка ]
Нервный гребень представляет собой группу временных мигрирующих клеток, которые остаются после нервной трубки закрытия ( нейруляции ), примерно на четвертой неделе эмбрионального развития. Они отвечают за дифференцировку в разнообразные группы клеток, которые достигают разных участков тела. Нервная трубка и нервный гребень происходят из эктодермы ; Нервная трубка продолжает формировать головной и спинной мозг , в то время как клетки нервного гребня в конечном итоге формируют различные кости и хрящи черепа и лица, мигрируя через глоточные дуги . Они также дифференцируются в сосудистую полоску улитки , нервы и глию кишечника ( мышечные сплетения ), шванновские клетки , которые миелинизируют периферическую нервную систему , чтобы обеспечить достаточную проводимость, одонтобласты , которые производят дентин глубоко в зубах, некоторые нейроэндокринные клетки. , соединительная ткань вокруг слюнных , слезных желез , гипофиза , тимуса и щитовидной желез, соединительная ткань глаза, например строма радужной оболочки. и роговица и трабекулярная сеть , [19] и меланоциты , в том числе те, что находятся в строме радужной оболочки, которые вызывают коричневый цвет глаз посредством меланина . Клетки нервного гребня также играют роль в формировании мышц, включая мышцы стенок некоторых сердечных артерий. [6]
Причины подтипов
[ редактировать ]- Тип 1 вызван аутосомно-доминантной мутацией гена PAX3 . [1] PAX3, или парный блок 3, представляет собой фактор транскрипции , который играет роль в поддержании открытого окна времени для определенных клеток нервного гребня (например, клеток головы и глаз) для деления и миграции до их терминальной дифференцировки (т. е. для поддержания их в состоянии стволовых клеток ). Таким образом, мутации в этом гене преждевременно останавливают их деление и миграцию, что приводит к незначительному отсутствию развития некоторых лицевых хрящей и костей, а также к недоразвитию структур внутреннего уха и отсутствию меланоцитов в строме радужной оболочки. Некоторые данные показывают, что PAX3 также регулирует клетки еще до формирования нервного гребня, т.е. нервной трубки, поскольку мыши с мутациями потери функции в одной из копий PAX3 имеют дефекты нервной трубки, такие как расщелина позвоночника или экзэнцефалия . [5]
- Тип 2 вызван мутацией любого из ряда генов, наиболее распространенным из которых является MITF , который классифицируется как тип 2А.
- Тип 2А вызван аутосомно-доминантной мутацией гена MITF . [7] MITF, или транскрипционный фактор, связанный с микрофтальмией, играет более специализированную роль в нервном гребне и более активно вовлекается после формирования нервного гребня (было обнаружено, что PAX3 и SOX10 активируют MITF ). [20] Известно, что он позволяет меланоцитам, остеокластам , тучным клеткам и клеткам пигментного эпителия сетчатки делиться и мигрировать. Участие остеокластов объясняет, почему мутации в обеих копиях MITF могут привести к упрочнению кости ( остепетрозу ), поскольку остеокласты ответственны за разрушение кости. MITF также активирует транскрипцию тирозиназы , фермента, выполняющего первый этап создания меланина (окисляющего тирозин ). Мутация в копии MITF также может привести к синдрому Титца , который отличается от синдрома Ваарденбурга равномерным альбинизмом вместо пятнистой депигментации. [5]
- Тип 2B вызван аутосомно-доминантной мутацией неизвестного гена на хромосоме 1 в диапазоне локусов 1p21–1p13.3. Ген был условно назван WS2B . [21] [22]
- Тип 2C вызван аутосомно-доминантной мутацией неизвестного гена на хромосоме 8 в локусе 8p23. Ген был условно назван WS2C . [23] [24]
- Тип 2D вызван аутосомно-рецессивной мутацией в обеих копиях гена SNAI2 . Исследование, обнаружившее эту связь, показало, что SNAI2 активируется MITF как часть развития нервного гребня, и это объясняет, почему мутации в MITF вызывают синдром Ваарденбурга, поскольку это приводит к отсутствию активации SNAI2 . мутации в единственной копии SNAI2 Также было обнаружено, что вызывают участки депигментации волос ( пегость ) без каких-либо других симптомов. [25]
- Тип 2E вызван аутосомно-доминантной мутацией гена SOX10 . [4]
- В редких случаях мутация в гене, отличном от известных в настоящее время, может быть ответственна за синдром Ваарденбурга с признаками типа 2. Обычно его первоначально классифицируют просто как тип 2, но ему может быть присвоен собственный подтип, как только ген или локус идентифицирован и установлен. . [7]
- Тип 3 вызван мутацией гена PAX3 , того же гена, что и при типе 1. [2] Он может наследоваться по аутосомно-доминантному или аутосомно-рецессивному типу; у двух родителей с синдромом Ваарденбурга 1-го типа может быть ребенок, несущий обе мутированные копии гена PAX3 (вероятность 25%) и имеющий синдром Ваарденбурга 3-го типа. миссенс-мутация Документально подтверждено, что имеет такой эффект. Однако синдром Ваарденбурга 3-го типа также может проявляться спонтанно только с одной мутированной копией PAX3 . Было документально подтверждено, что удаление парного домена гена приводит к такому эффекту. [26] [5] Однако существенной корреляции между типом мутации и тяжестью заболевания обнаружено не было. Тяжесть, как правило, определяется мутациями в других генах ( эпистаз ), о чем свидетельствуют отчетливые семейные закономерности тяжести, не связанные с типом мутации Ваарденбурга. [5] Мутации в обеих копиях PAX3 иногда приводили к смерти до или вскоре после рождения, а мыши с мутациями потери функции в обеих копиях гена не выживают. [5]
- Тип 4 вызван мутацией любого из ряда генов, наиболее распространенным из которых является SOX10 , который классифицируется как тип 4C.
Исследование было проведено на редком случае двойной гетерозиготы у ребенка, у которого у каждого родителя были только единичные мутации в MITF или PAX3 . Эффект двойных гетерозиготных мутаций в генах MITF и PAX3 при WS1 и WS2 может усиливать симптомы, связанные с пигментацией. Это приводит к выводу, что двойная мутация MITF связана с конечной формой синдрома Ваарденбурга и может влиять на фенотипы или симптомы синдрома. [28]
Таблица классификации
[ редактировать ]| Тип | МОЙ БОГ | Ген | Локус | Наследование |
|---|---|---|---|---|
| Тип 1 (WS1) | 193500 | ПАКС3 | 2q36.1 [29] | Аутосомно-доминантный |
| Тип 2А (WS2A, первоначально WS2) | 193510 | МИТФ | 3p14.1–p12.3 | Аутосомно-доминантный |
| Тип 2Б (ВС2Б) | 600193 | WS2B | 1p21–p13.3 | Аутосомно-доминантный |
| Тип 2C (WS2C) | 606662 | WS2C | 8p23 | Аутосомно-доминантный |
| Тип 2D (WS2D) | 608890 | СНАИ2 | 8q11 | Аутосомно-рецессивный |
| Тип 2Е (ВС2Е) | 611584 | SOX10 | 22q13.1 | Аутосомно-доминантный |
| Тип 3 (WS3) | 148820 | ПАКС3 | 2q36.1 | Аутосомно-доминантный или аутосомно-рецессивный |
| Тип 4А (WS4A) | 277580 | ЕДНРБ | 13q22 | Аутосомно-доминантный или аутосомно-рецессивный |
| Тип 4Б (WS4B) | 613265 | ЭДН3 | 20q13 | Аутосомно-доминантный или аутосомно-рецессивный |
| Тип 4С (ВС4К) | 613266 | SOX10 | 22q13.1 | Аутосомно-доминантный |
Уход
[ редактировать ]В настоящее время не существует лечения синдрома Ваарденбурга. Симптомом, который, скорее всего, будет иметь практическое значение, является глухота, и ее лечат так же, как и любую другую необратимую глухоту. В отмеченных случаях могут возникнуть косметические проблемы. Другие нарушения (неврологические, структурные, болезнь Гиршпрунга), связанные с синдромом, лечатся симптоматически.
Эпидемиология
[ редактировать ]Распространенность всех типов синдрома Ваарденбурга оценивается в 1 на 42 000. [5] [8] Типы 1 и 2 являются наиболее распространенными, а тип 1 встречается немного чаще. [30] [31] В обзоре 2015 года, в котором приняли участие 417 пациентов, тип 1 оказался наиболее распространенным типом, охватывающим около половины всех случаев (47%), тогда как тип 2 был вторым наиболее распространенным типом, охватывающим около трети (33%) . [8] Подавляющее большинство (около 85%) случаев типа 2 относятся к типу 2А. [8] Распространенность типа 2В неизвестна, поскольку о ней сообщалось только в одном исследовании 1996 года. [22] Тип 2C пока обнаружен только в одной итальянской семье. [23] [24] а тип 2D был обнаружен только у 2 неродственных пациентов по состоянию на 2018 год. [update]. [13] [8] [32] Сообщается, что по состоянию на 2017 год количество известных случаев типа 2E, сопровождающихся неврологическими нарушениями, составило 23. [update], [33] а количество остальных неизвестно. Тип 3 встречается реже, чем типы 1, 2 и 4. [34] составляет менее 2% случаев. [8] Тип 4, по-видимому, охватывает примерно пятую часть случаев (19%). Из его подтипов тип 4C является наиболее распространенным (около 71% от типа 4), за ним следуют тип 4A (19%) и тип 4B (10%). [8]
Подсчитано, что синдром Ваарденбурга присутствует у 2–5% врожденно глухих людей. Врожденная глухота составляет около половины глухоты в целом. [8] Около 1 из 30 учащихся школ для глухих страдает синдромом Ваарденбурга. Изменчивость проявлений синдрома затрудняет получение точных данных о его распространенности. [8]
История
[ редактировать ]Ранние описания
[ редактировать ]В 1916 году голландский офтальмолог Ян ван дер Хуве (1878–1952) описал пару девочек-близнецов с глухотой и особым типом блефарофимоза , предположительно являющегося канторной дистопией, встречающейся при синдроме Ваарденбурга типов 1 и 3. [8] [35] Блефарофимоз описывает веки, которые недоразвиты и постоянно закрывают часть глаз.
В 1926 году немецкий врач Ирмгард Менде описала семью из четырех поколений, в которой у пятерых детей были симптомы депигментации волос, кожи и глаз, глухота и « монголоидная » внешность. (Позже Ваарденбург приписал это описание антиутопии канторума.) [36] [35] Позже это привело к тому, что в некоторых базах данных был зафиксирован синоним синдрома Менде. [11] [37]
В 1929 году голландский врач К.Т.А. Хальбертсма описал семейную модель антиутопии канторум. [38] [35] а в 1930 году итальянский врач Винченцо Гуальди. [39] (1891–1976) также подтвердили наследственный характер дистопии канторум. [36] Позже это привело к тому, что в некоторых базах данных был зарегистрирован синоним синдрома Ван дер Хуве-Хальбертсма-Ваарденбурга-Гуальди. [11]
В 1947 году швейцарский офтальмолог Дэвид Кляйн (1908–1993) впервые сообщил о пациенте с двусторонней глухотой, нарушениями пигментации, характерными чертами лица и пороками развития рук. Хотя это было первое полное описание пациента с синдромом Ваарденбурга 3-го типа, современные клиницисты не считали синдром, который он описал, таким же, как синдром, описанный Ваарденбургом четыре года спустя, отчасти из-за того, насколько серьезными были пороки развития рук у него. пациент. [40]
Синдром был впервые полностью формализован и описан голландским офтальмологом и генетиком Петрусом Йоханнесом Ваарденбургом (1886–1979) в 1951 году. [9] [10] Состояние, которое он описал, теперь классифицируется как синдром Ваарденбурга 1-го типа.
Описания подтипов
[ редактировать ]Тип 2 был впервые установлен в 1971 году, когда исследование показало, что у некоторых пациентов с синдромом Ваарденбурга не было канторной дистопии. [7] [41] Исследование 1977 года подтвердило семейную модель этого другого проявления. [7] Два исследования 1994 года впервые подтвердили связь между этим типом синдрома Ваарденбурга и мутациями в гене MITF (теперь классифицированном как тип 2А), расположенном на хромосоме 3 в локусе 3p14.1–p12.3. [7]
Тип 2B был впервые установлен в 1994 году, когда в том же исследовании, которое выявило мутации в MITF у пациентов с синдромом Ваарденбурга 2 типа, также было обнаружено, что у некоторых пациентов не было никаких мутаций в этом регионе. [21] [42] Второе исследование 1994 года обнаружило связь с хромосомой 1 в локусе 1p21–p13.3. Это состояние стало известно как тип 2B (с геном, обозначенным WS2B ), однако с тех пор оно не было документировано, и ответственный за него ген остается неизвестным. [21] [22]
Тип 2C был установлен в 2001 году, когда исследование итальянской семьи с признаками синдрома Ваарденбурга 2 типа показало, что они обусловлены неизвестным геном на хромосоме 8 в локусе 8q23, который был нарушен хромосомной транслокацией . В ходе исследования было установлено предварительное название гена — WS2C . Однако мутации в этой области у пациентов с синдромом Ваарденбурга с тех пор не обнаруживались. [23] [24]
Тип 2D был установлен в 2002 году, когда исследование, направленное на поиск мутаций в человеческой версии гена SNAI2 , который, как известно, вызывает депигментацию у мышей, обнаружило делеции обеих копий этого гена у двух неродственных людей с синдромом Ваарденбурга 2 типа. Мутации у обоих С тех пор копии этого гена не были обнаружены у пациентов с синдромом Ваарденбурга 2 типа. [8]
Тип 2E был впервые установлен в 1996 году, когда в ходе исследования была выявлена девочка с симптомами синдрома Ваарденбурга 2-го типа, но с дополнительным недоразвитием передней части глаза , что привело к слепоте. В 1999 году было обнаружено, что у нее была мутация в гене SOX10 , а более поздние исследования подтвердили связь между мутациями этого гена и этим фенотипом, а также неврологическими симптомами, такими как задержка развития. [4]
Тип 3 впервые получил свое название от Goodman et al. в 1981 году в сотрудничестве с Кляйн, в ходе которого они установили связь с аномалиями рук, о которых впервые сообщил Кляйн в 1947 году. [40] Мутации PAX3 впервые были связаны с этим фенотипом в 1992 году. [2]
Коморбидность с болезнью Гиршпрунга, которая позже составит 4 тип, впервые была отмечена в различных исследованиях в 1970-х годах. Индийский педиатр Кришнакумар Шах и его коллеги впервые описали этот синдром как возможный вариант синдрома Ваарденбурга в 1981 году. [43] Этот вариант впервые был объяснен мутацией EDNRB в 1994 году (сейчас классифицируется как тип 4А). [3] Тип 4B был установлен в 1996 году, когда было обнаружено, что мутации в EDN3 приводят к этому типу синдрома Ваарденбурга. [27] а тип 4C был впервые установлен в 1998 году, когда было обнаружено, что мутации в SOX10 также приводят к этому типу. [16]
Общество и культура
[ редактировать ]Популярная культура
[ редактировать ]- В романе Шок « Робина Кука » 2001 года упоминается персонаж, страдающий этим расстройством. [44]
- Энцо Маклауд, главный герой Питера Мэя серии книг «Досье Энцо» 2006–2017 годов , страдает синдромом Ваарденбурга. Его глаза разного цвета, а в волосах белая прядь. [45] [46]
- В шестого сезона эпизоде 2011 года сериала «Кости» «Знаки в тишине» команда должна раскрыть дело, в котором у подозреваемого убийцы синдром Ваарденбурга. [47]
- В книге Кимберли МакКрайт «Реконструкция Амелии» 2013 года персонажей представлены несколько с симптомами Ваарденбурга. [48]
- В книге «Ближе, чем вы думаете» 2014 Карен Роуз года представлены три персонажа, братья и сестры, с синдромом Ваарденбурга. [49]
- В книге Убийство в храме майя» 2017 года фигурируют несколько персонажей с синдромом Ваарденбурга. Эм. Дж. Мандрагоры « [50] [ нужен лучший источник ]
- В романе Чендлера Бейкера «Сеть шепота» 2019 года этот синдром используется как сюжет. [ нужна ссылка ]
Известные люди
[ редактировать ]- У канадского YouTube-блогера Стефа Санджати синдром Ваарденбурга 1-го типа. [51]
Другие животные
[ редактировать ]
Синдром Ваарденбурга типа 2А (с мутацией в MITF ) обнаружен у собак, крупного рогатого скота Флекви , норок , мышей и золотистого хомячка . [52] Дегенерация улитки и мешочка , наблюдаемая при синдроме Ваарденбурга, также обнаружена у глухих белых кошек, далматинов и собак других пород, белых норок и мышей. [53]
Домашние кошки с голубыми глазами и белой шерстью часто бывают совершенно глухими. [54] Глухота гораздо чаще встречается у белых кошек, чем у кошек других окрасов шерсти. Согласно « » ASPCA Полному руководству по кошкам , «от 17 до 20 процентов белых кошек с неголубыми глазами глухие; 40 процентов белых кошек с «разноглазыми» глазами и одним голубым глазом глухие; и от 65 до 85 процентов голубоглазых кошек. Белоглазые кошки глухи». [55] Хотя было проведено мало исследований, чтобы связать это с генами, которые, как известно, участвуют в синдроме Ваарденбурга у человека, генетическое нарушение развития нервного гребня может привести к такому проявлению и у кошек. [56] Один из генов, который при мутации приводит к глухоте и белой шерсти у кошек, KIT . [57] Было обнаружено, что он увеличивает экспрессию MITF . [58]
Летальный белый синдром — синдром у лошадей, вызванный мутациями в обеих копиях EDNRB . Это приводит к смерти от кишечной псевдонепроходимости вследствие болезни Гиршпрунга. Однако мутация в единственной копии EDNRB , как и при синдроме Ваарденбурга типа 4А, приводит к образованию пятнистой белой шерсти оверо с глухотой. [59]
У хорьков с синдромом Ваарденбурга есть небольшая белая полоса вдоль макушки или затылка, а иногда и вдоль задней части шеи (так называемая «блестящая» шерсть) или сплошная белая полоса на голове от носа до плеч (так называемая «блестящая» полоса на голове). узор шерсти «панда»). Больные хорьки часто имеют более плоский череп и более широко посаженные глаза, чем здоровые хорьки. Поскольку у здоровых хорьков плохой слух, глухоту можно обнаружить только по отсутствию реакции на громкие звуки. Поскольку это наследственное заболевание, больных животных нельзя использовать для разведения. Исследование корреляции между вариациями шерсти и глухотой у европейских хорьков показало: «Все (n = 27) панды, американские панды и пламенные хорьки были глухими». [60]
См. также
[ редактировать ]- Синдром Чедиака-Хигаси , аналогичный синдром, включающий иммунодефицит и периферическую невропатию.
- Синдром Титца , состояние, подобное синдрому Ваарденбурга 2-го типа, связанное с однородным альбинизмом (вызванное мутациями в MITF ).
- Болезнь Фогта-Коянаги-Харады , аутоиммунное заболевание, вызывающее увеит, очаговую депигментацию и симптомы внутреннего уха.
Ссылки
[ редактировать ]- ^ Jump up to: а б с д «Запись OMIM — # 193500 — СИНДРОМ ВАРДЕНБУРГА, ТИП 1; WS1» . omim.org . Проверено 7 декабря 2019 г.
- ^ Jump up to: а б с д и «Запись OMIM — # 148820 — СИНДРОМ ВАРДЕНБУРГА, ТИП 3; WS3» . omim.org . Проверено 7 декабря 2019 г.
- ^ Jump up to: а б с д «Запись OMIM — # 277580 — СИНДРОМ ВАРДЕНБУРГА, ТИП 4A; WS4A» . omim.org . Проверено 7 декабря 2019 г.
- ^ Jump up to: а б с д «Запись OMIM — # 611584 — СИНДРОМ ВАРДЕНБУРГА, ТИП 2E; WS2E» . omim.org . Проверено 7 декабря 2019 г.
- ^ Jump up to: а б с д и ж г час Пинго, Вероника; Энте, Доротея; Моал, Флоренс Дасто-Ле; Гуссенс, Мишель; Марлин, Сандрин; Бондюран, Надеж (2010). «Обзор и обновление мутаций, вызывающих синдром Ваарденбурга» . Человеческая мутация . 31 (4): 391–406. дои : 10.1002/humu.21211 . ISSN 1098-1004 . ПМИД 20127975 . S2CID 12278025 .
- ^ Jump up to: а б «Развитие нервного гребня – эмбриология» . embreology.med.unsw.edu.au . Проверено 13 декабря 2019 г.
- ^ Jump up to: а б с д и ж г час «Запись OMIM — # 193510 — СИНДРОМ ВАРДЕНБУРГА, ТИП 2A; WS2A» . omim.org . Проверено 7 декабря 2019 г.
- ^ Jump up to: а б с д и ж г час я дж к л м н Сонг, Дж.; Фэн, Ю.; Акке, Франция; Кук, П.; Влеминкс, К.; Дхуге, Эй Джей (2016). «Потеря слуха при синдроме Ваарденбурга: систематический обзор». Клиническая генетика . 89 (4): 416–425. дои : 10.1111/cge.12631 . ISSN 1399-0004 . ПМИД 26100139 . S2CID 23834634 .
- ^ Jump up to: а б Чандра Мохан, Сетти. ЛН (01.09.2018). «Случай синдрома Ваарденбург-Шаха в семье с обзором литературы» . Журнал отологии . 13 (3): 105–110. дои : 10.1016/j.joto.2018.05.005 . ISSN 1672-2930 . ПМК 6291636 . ПМИД 30559775 .
- ^ Jump up to: а б Ваарденбург П.Дж. (сентябрь 1951 г.). аномалии развития век, бровей и корня носа с пигментными аномалиями радужной оболочки и волос на голове и с врожденной ограниченной глухотой Новый синдром, сочетающий (лейкизм, полиоз) и врожденной глухотой (глухотой) » . Являюсь. Дж. Хум. Жене 3 (3): 195–253. ПМК 1716407 . ПМИД 14902764 .
- ^ Jump up to: а б с «Синдром Ваарденбурга | Информационный центр по генетическим и редким заболеваниям (GARD) – программа NCATS» . Rarediseases.info.nih.gov . Проверено 17 апреля 2018 г.
- ^ «Синдром Ваарденбурга» . Домашний справочник по генетике . Октябрь 2012.
- ^ Jump up to: а б «Запись OMIM — # 608890 — СИНДРОМ ВАРДЕНБУРГА, ТИП 2D; WS2D» . omim.org . Проверено 7 декабря 2019 г.
- ^ Барал В., Чауи А., Ватанабэ И., Гуссенс М., Атти-Битач Т., Марлин С., Пинго В., Бондюран Н. (2012). «Скрининг регуляторных областей MITF и SOX10 при синдроме Ваарденбурга 2 типа» . ПЛОС ОДИН 7 (7): е41927. Бибкод : 2012PLoSO... 741927B дои : 10.1371/journal.pone.0041927 . ПМК 3407046 . ПМИД 22848661 .
- ^ Jump up to: а б Пирпон, JW; Сен-Жак, Д.; Сивер, Л.Х.; Эриксон, Р.П. (март 1995 г.). «Семья с необычным синдромом Ваарденбурга I типа (WSI), расщелиной губы (неба) и болезнью Гиршпрунга не связана с PAX 3». Клиническая генетика . 47 (3): 139–143. дои : 10.1111/j.1399-0004.1995.tb03946.x . ISSN 0009-9163 . ПМИД 7634536 . S2CID 24530898 .
- ^ Jump up to: а б с «Запись OMIM — # 613266 — СИНДРОМ ВАРДЕНБУРГА, ТИП 4C; WS4C» . omim.org . Проверено 7 декабря 2019 г.
- ^ «Сирота: В поисках болезни» . www.orpha.net . Проверено 10 декабря 2019 г.
- ^ Верхей, Йоханна БГМ; Сивал, Дебора А.; ван дер Хувен, Йоханнес Х.; Вос, Ивонн Дж.; Майнерс, Линда С.; Брауэр, Обеле Ф.; ван Эссен, Энтони Дж. (январь 2006 г.). «Синдром Шаха-Ваарденбурга и PCWH, связанный с мутациями SOX10: описание случая и обзор литературы». Европейский журнал детской неврологии . 10 (1): 11–17. дои : 10.1016/j.ejpn.2005.10.004 . ISSN 1090-3798 . ПМИД 16504559 .
- ^ Уильямс, Антионетт Л.; Бонсак, Бренда Л. (июнь 2015 г.). «Производные нервного гребня в развитии глаз: распознавание ока бури» . Исследование врожденных дефектов, часть C: Эмбрион сегодня: обзоры . 105 (2): 87–95. дои : 10.1002/bdrc.21095 . ISSN 1542-975Х . ПМК 5262495 . ПМИД 26043871 .
- ^ Каваками, Акинори; Фишер, Дэвид Э. (июнь 2017 г.). «Основная роль фактора транскрипции, связанного с микрофтальмией, в биологии меланоцитов и меланомы» . Лабораторные исследования; Журнал технических методов и патологии . 97 (6): 649–656. дои : 10.1038/labinvest.2017.9 . ISSN 1530-0307 . ПМИД 28263292 .
- ^ Jump up to: а б с «Запись OMIM — % 600193 — СИНДРОМ ВАРДЕНБУРГА, ТИП 2B; WS2B» . www.omim.org . Проверено 23 декабря 2019 г.
- ^ Jump up to: а б с Лалвани, АК; Сан-Агустин, ТБ; Уилкокс, скорая помощь (1 сентября 1994 г.). «Локус синдрома Ваарденбурга типа II картируется на хромосоме 1p13.3-2.1». Американский журнал генетики человека . 55 (Приложение 3). ОСТИ 133315 .
- ^ Jump up to: а б с «Запись OMIM — % 606662 — СИНДРОМ ВАРДЕНБУРГА, ТИП 2C; WS2C» . omim.org . Проверено 7 декабря 2019 г.
- ^ Jump up to: а б с Селикорни, Анджело; Гернери, Сильвана; Ратти, Антония; Пиццути, Антонио (январь 2002 г.). «Цитогенетическое картирование нового локуса синдрома Ваарденбурга II типа». Генетика человека . 110 (1): 64–67. дои : 10.1007/s00439-001-0643-9 . ISSN 0340-6717 . ПМИД 11810298 . S2CID 24411957 .
- ^ Санчес-Мартин, Мануэль; Перес-Лосада, Хесус; Родригес-Гарсия, Аранча; Гонсалес-Санчес, Белен; Корф, Брюс Р.; Кастер, В.; Мосс, Селия; Спритц, Ричард А.; Санчес-Гарсия, И. (2003). «Удаление гена SLUG (SNAI2) приводит к пегисти человека». Американский журнал медицинской генетики, часть A. 122А (2): 125–132. дои : 10.1002/ajmg.a.20345 . ISSN 1552-4833 . ПМИД 12955764 . S2CID 33811699 .
- ^ Текин, М.; Бодурта, JN; Нэнси, МЫ; Пандия, А. (2001). «Синдром Ваарденбурга типа 3 (синдром Клейна-Ваарденбурга) с сегрегацией с гетерозиготной делецией в парном боксерском домене PAX3: простой вариант или истинный синдром?». Клиническая генетика . 60 (4): 301–304. дои : 10.1034/j.1399-0004.2001.600408.x . ISSN 1399-0004 . ПМИД 11683776 . S2CID 24025063 .
- ^ Jump up to: а б «Запись OMIM — # 613265 — СИНДРОМ ВАРДЕНБУРГА, ТИП 4B; WS4B» . omim.org . Проверено 7 декабря 2019 г.
- ^ Ян Т, Ли X, Хуан Q, Ли L, Чай Y, Сунь Л, Ван X, Чжу Y, Ван Z, Хуан Z, Ли Y, Ву Х (январь 2013 г.). «Двойные гетерозиготные мутации MITF и PAX3 приводят к синдрому Ваарденбурга с повышенной пенетрантностью пигментных дефектов» . Клин. Жене . 83 (1): 78–82. дои : 10.1111/j.1399-0004.2012.01853.x . ПМИД 22320238 . S2CID 34173541 .
- ^ Цукамото К., Накамура Й., Ниикава Н. (март 1994 г.). «Выделение двух изоформ транскриптов гена PAX3 и их тканеспецифичная альтернативная экспрессия в тканях взрослого человека». Хм. Жене . 93 (3): 270–4. дои : 10.1007/bf00212021 . ПМИД 7545913 . S2CID 36749688 .
- ^ «Орфанет: синдром Ваарденбурга 1 типа» . www.orpha.net . Проверено 10 декабря 2019 г.
- ^ «Синдром Ваарденбурга типа II» (PDF) . Сирота . 2005. Архивировано из оригинала (PDF) 24 апреля 2021 года . Проверено 10 декабря 2019 г.
- ^ Салим, Мохаммед Д. (2019). «Биология развития меланоцитов человека, пьебальдизм и синдром Ваарденбурга». Детская дерматология . 36 (1): 72–84. дои : 10.1111/pde.13713 . ISSN 1525-1470 . ПМИД 30561083 .
- ^ Богданова-Михайлова, Петя; Александр, Майкл Д.; Мерфи, Рэймонд П.Дж.; Мерфи, Шинеад М. (2017). «Синдром Ваарденбурга: редкая причина наследственной невропатии из-за мутации SOX10». Журнал периферической нервной системы . 22 (3): 219–223. дои : 10.1111/jns.12221 . ISSN 1529-8027 . ПМИД 28544110 . S2CID 13676694 .
- ^ «Орфанет: синдром Ваарденбурга 3 типа» . www.orpha.net . Проверено 10 декабря 2019 г.
- ^ Jump up to: а б с Де Хаас, EBH; Тан, KEWP (1 января 1966 г.). «Синдром Ваарденбурга». Документа Офтальмологическая . 21 (1): 239–282. дои : 10.1007/BF00184136 . ISSN 1573-2622 . S2CID 10163905 .
Ваарденбург (1951, 1957, 1961) высказал мнение, что все эти случаи неосложненного блефарофимоза действительно относятся к его синдрому и что этот тип канторной дистопии не встречается как отдельный признак. ... Что касается случаев Менде, [Ваарденбург] считает, что «монголоидный компонент» у этих пациентов на самом деле был вызван дистопией канторум.
- ^ Jump up to: а б Кляйн, Д. (февраль 1983 г.). «Историческая основа и доказательства доминантного наследования синдрома Клейна-Ваарденбурга (тип III)». Американский журнал медицинской генетики . 14 (2): 231–239. дои : 10.1002/ajmg.1320140205 . ISSN 0148-7299 . ПМИД 6340503 .
- ^ Стедман, Томас Латроп (2005). Медицинские эпонимы Стедмана . Липпинкотт Уильямс и Уилкинс. ISBN 978-0-7817-5443-9 .
- ^ «О двух схожих, но существенно различных врожденных дефектах глаз» . Голландский медицинский журнал (на голландском языке). 02.12.2009 . Проверено 10 декабря 2019 г.
- ^ Ежегодник Министерства национального образования (на итальянском языке). Главное управление штата. 1935. с. 142.
- ^ Jump up to: а б Гудман, Р.М.; Левиталь, И.; Соломон, А.; Кляйн, Д. (апрель 1982 г.). «Поражение верхних конечностей при синдроме Кляйна-Ваарденбурга». Американский журнал медицинской генетики . 11 (4): 425–433. дои : 10.1002/ajmg.1320110407 . ISSN 0148-7299 . ПМИД 7091186 .
- ^ Ариас, С. (март 1971 г.). «Генетическая гетерогенность при синдроме Ваарденбурга». Оригинальная серия статей о врожденных дефектах . 07 (4): 87–101. ISSN 0547-6844 . ПМИД 5006208 .
- ^ Хьюз, А.Е.; Ньютон, Вирджиния; Лю, XZ; Рид, AP (август 1994 г.). «Ген синдрома Ваарденбурга 2 типа картируется близко к человеческому гомологу гена микрофтальмии на хромосоме 3p12-p14.1». Природная генетика . 7 (4): 509–512. дои : 10.1038/ng0894-509 . ISSN 1061-4036 . ПМИД 7951321 . S2CID 2913481 .
- ^ Шах, Кришнакумар Н.; Далал, Субхаш Дж.; Десаи, Мина П.; Шет, Прем Н.; Джоши, Нана С.; Амбани, Лалит М. (1 сентября 1981 г.). «Белый чуб, нарушение пигментации радужной оболочки и болезнь Гиршпрунга длинного сегмента: возможный вариант синдрома Ваарденбурга». Журнал педиатрии . 99 (3): 432–435. дои : 10.1016/S0022-3476(81)80339-3 . ISSN 0022-3476 . ПМИД 7264803 .
- ^ Кук, Робин (12 декабря 2011 г.). Шок . Пан Макмиллан. п. 175. ИСБН 978-1-4472-1796-1 .
- ^ Мэй, Питер (30 мая 2013 г.). Необыкновенные люди: Энцо Маклауд 1 . Кверкус. п. 32. ISBN 978-1-78206-885-3 .
- ^ Мэй, Питер (13 июня 2013 г.). Черный свет синий: Энцо Маклауд 3 . Кверкус. п. 76. ИСБН 978-1-78206-887-7 .
- ^ «Резюме Bones 6.21 «Знаки в тишине» - журнал Persephone» . Архивировано из оригинала 11 октября 2011 г. Проверено 14 декабря 2019 г.
- ^ МакКрайт, Кимберли (01 апреля 2013 г.). Реконструкция Амелии . Саймон и Шустер. ISBN 978-1-4711-2944-5 .
- ^ Роуз, Карен (6 ноября 2014 г.). Ближе, чем вы думаете (Цинциннати, книга 1) . Заголовок. ISBN 978-0-7553-8999-5 .
- ^ минони. «Знакомьтесь, хороший сыщик» . Amazon.com . Проверено 14 декабря 2019 г.
- ^ Эдвардс, Люси (2018). « Я понимаю, ты трансгендер, но что у тебя с лицом?» " . Новости Би-би-си . Проверено 28 января 2018 г.
- ^ МАРКАКИС, МАРИОС Н.; СЕДРИНГ, VIWEEK E.; ДАНЦЕР, ВИБЕКЕ; КРИСТЕНСЕН, КНУД; АНИСТОРОАЕЙ, РАЗВАН (01.08.2014). «Ассоциация гена MITF с фенотипом слуха и пигментации у белой американской норки Хедлунд (Neovison vison)» Журнал генетики . 93 (2): 477–481. дои : 10.1007/ s12041-014-0370-3 hdl : 10067/1211550151162165141 . ISSN 0973-7731 . ПМИД 25189243 . S2CID 16725018 .
- ^ Стрейн, Джордж М. (2015). «Генетика глухоты домашних животных» . Границы ветеринарной науки . 2:29 . дои : 10.3389/fvets.2015.00029 . ISSN 2297-1769 . ПМЦ 4672198 . ПМИД 26664958 .
- ^ Уэбб А.А., Каллен К.Л. (июнь 2010 г.). «Цвет шерсти и связанные с окрасом шерсти неврологические и нейроофтальмологические заболевания» . Может. Ветеринар. Дж . 51 (6): 653–7. ПМЦ 2871368 . ПМИД 20808581 .
- ^ Ричардс Дж (1999). Полное руководство ASPCA по кошкам: все, что вам нужно знать о выборе домашнего питомца и уходе за ним . Книги летописи. п. 71. ИСБН 9780811819299 .
- ^ Оменн, Гилберт С.; МакКьюсик, Виктор А.; Горлин, Роберт Дж. (1979). «Ассоциация синдрома Ваарденбурга и мегаколона Гиршпрунга». Американский журнал медицинской генетики . 3 (3): 217–223. дои : 10.1002/ajmg.1320030302 . ISSN 1096-8628 . ПМИД 484594 .
Глухая, голубоглазая белая кошка, отмеченная Бри [1829] и Дарвином [1892] и изученная гистологически на рубеже веков [Александр, 1900], имеет вариабельную клиническую и гистологическую картину, обусловленную одним из двух аутосомно-доминантные гены... Эти плейотропные эффекты отдельных генов можно объяснить воздействием на клетки нервного гребня, участвующие в возникновении всех тканей, пораженных синдромом Ваарденбурга [Weston, 1969].
- ^ Дэвид, Виктор А.; Менотти-Рэймонд, Мэрилин; Уоллес, Андреа Кутс; Ролке, Мелоди; Келер, Джеймс; Лейти, Роберт; Эйзирик, Эдуардо; Ханна, Стивен С.; Нельсон, Джордж; Шеффер, Алехандро А.; Коннелли, Кэтрин Дж. (01 октября 2014 г.). «Вставка эндогенного ретровируса в онкоген KIT определяет белую и белую пятнистость у домашних кошек» . G3: Гены, геномы, генетика . 4 (10): 1881–1891. дои : 10.1534/g3.114.013425 . ISSN 2160-1836 . ПМК 4199695 . ПМИД 25085922 .
- ^ Ли, Юл-Нам; Брандал, Стефани; Ноэль, Пьер; Вентцель, Эрик; Менделл, Джошуа Т.; Макдевитт, Майкл А.; Капур, Рубен; Картер, Мелоди; Меткалф, Дин Д.; Такемото, Клиффорд М. (31 марта 2011 г.). «Передача сигналов KIT регулирует экспрессию MITF через микроРНК при пролиферации нормальных и злокачественных тучных клеток» . Кровь . 117 (13): 3629–3640. дои : 10.1182/blood-2010-07-293548 . ISSN 0006-4971 . ПМК 3072881 . ПМИД 21273305 .
- ^ Магдезиан, К. Гэри; Уильямс, Д. Колетт; Алеман, Моника; ЛеКутер, Ричард А.; Мэдиган, Джон Э. (15 ноября 2009 г.). «Оценка глухоты у американских пейнтбольных лошадей по фенотипу, слуховым реакциям ствола мозга и генотипу рецептора B эндотелина» . Журнал Американской ветеринарной медицинской ассоциации . 235 (10): 1204–1211. дои : 10.2460/javma.235.10.1204 . ISSN 0003-1488 . ПМИД 19912043 .
- ^ Пьяцца С., Абитболь М., Гнирс К., Хюн М., Козиниль Л. (май 2014 г.). «Распространенность глухоты и ее связь с вариациями шерсти у хорьков, принадлежащих клиентам» . Дж. Ам. Ветеринар. Мед. доц . 244 (9): 1047–52. дои : 10.2460/javma.244.9.1047 . ПМИД 24739114 .
Внешние ссылки
[ редактировать ]- Запись GeneReviews/NCBI/NIH/UW о синдроме Ваарденбурга типа I
- Каталог генетических заболеваний OMIM — синдром Ваарденбурга
| Базы данных органов управления : Национальные |
|---|