синдром Пфайфера
| синдром Пфайфера | |
|---|---|
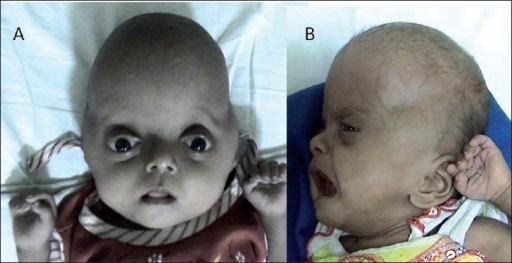 | |
| Синдром Пфайффера 2 типа с черепом в форме клеверного листа и двусторонним проптозом до и после операции | |
| Специальность | Ревматология |
| Причины | Генетический [1] |
| Частота | 1 на 100 000 рождений [1] |
| Назван в честь | Рудольф Артур Пфайффер |
Синдром Пфайффера — редкое генетическое заболевание , характеризующееся преждевременным сращением определенных костей черепа ( краниосиностоз ) , что влияет на форму головы и лица. Синдром включает в себя аномалии рук и ног, такие как широкие и отклоненные большие пальцы рук и ног.
Синдром Пфайффера вызван мутациями рецепторов фактора роста фибробластов FGFR1 и FGFR2 . Синдром разделен на три типа: тип 1 (классический синдром Пфайффера) более мягкий и вызван мутациями в любом гене; типы 2 и 3 являются более тяжелыми и часто приводят к смерти в младенчестве, вызванной мутациями FGFR2 . [2]
Лечения синдрома не существует. Лечение носит поддерживающий характер и часто включает хирургическое вмешательство в первые годы жизни для коррекции деформаций черепа и дыхательной функции. [2] Большинство людей с синдромом Пфайффера 1-го типа имеют нормальный интеллект и продолжительность жизни; типы 2 и 3 обычно вызывают нарушения нервного развития и раннюю смерть. В более позднем возрасте хирургическое вмешательство может помочь в формировании костей и строении лица.
Синдром Пфайффера встречается примерно у 1 из 100 000 человек. [1] Синдром назван в честь немецкого генетика Рудольфа Артура Пфайффера (1931–2012), описавшего его в 1964 году. [3]
Признаки и симптомы
[ редактировать ]

Многие черты лица являются результатом преждевременного сращения костей черепа ( краниосиностоз ). Голова не может нормально расти, что приводит к высокому и выступающему лбу ( турри -брахицефалия ) и глазам, которые кажутся выпуклыми ( проптоз ) и широко посаженными ( гипертелоризм ). Кроме того, имеется недоразвитая верхняя челюсть ( верхнечелюстная гипоплазия ). Более половины детей с синдромом Пфайффера страдают потерей слуха; проблемы с зубами являются обычным явлением. [4] У ребенка с синдромом Пфайффера может быть маленький нос в форме клюва; скученные, кривые зубы; и апноэ во сне из-за заложенности носа. Существует три основных типа синдрома Пфайффера: тип I — самый легкий и наиболее распространенный; тип II является наиболее тяжелым, с неврологическими проблемами и деформацией клеверного листа; и тип III подобен типу II, но без деформации клеверного листа. [5]
У людей с синдромом Пфайффера большие пальцы и первые (большие) пальцы ног широкие и отгибаются от других пальцев ( варус большого пальца стопы и варус большого пальца стопы ). Также распространены необычно короткие пальцы рук и ног ( брахидактилия ), а между пальцами могут быть перепонки или сращения ( синдактилия ). [6]
Причина
[ редактировать ]Синдром Пфайффера тесно связан с мутациями рецептора 1 фактора роста фибробластов ( FGFR1 ) на хромосоме 8 или гена рецептора 2 фактора роста фибробластов ( FGFR2 ) на хромосоме 10 . [1] [7] [8] [9] Эти гены кодируют рецепторы фактора роста фибробластов , которые важны для нормального развития костей. [10] Считается, что преклонный возраст отца является фактором риска спорадических случаев синдрома Пфайффера из-за увеличения мутаций в сперме по мере того, как мужчины становятся старше. [1] [11]
Диагностика
[ редактировать ]Классификация
[ редактировать ]Наиболее широко принятая клиническая классификация синдрома Пфайффера была опубликована М. Майклом Коэном в 1993 году. [1] [12] Коэн разделил синдром на три, возможно, перекрывающихся типа, каждый из которых характеризуется широкими большими пальцами рук, широкими большими пальцами ног, брахидактилией и, возможно, синдактилией : [13]
- Тип 1, также известный как классический синдром Пфайффера, включает краниосиностоз и «дефицит средней зоны лица». Этот тип наследуется по аутосомно-доминантному типу. Большинство людей с синдромом Пфайффера 1 типа имеют нормальный интеллект и нормальную продолжительность жизни.
- Тип 2 включает череп в форме клеверного листа из-за обширного сращения костей, а также тяжелого проптоза . Этот тип встречается спорадически (т.е., по-видимому, не передается по наследству) и имеет «плохой прогноз и тяжелые неврологические нарушения, обычно с ранней смертью».
- Тип 3 включает краниосиностоз и тяжелый экзофтальм. Этот тип возникает спорадически (т.е., по-видимому, не передается по наследству) и имеет «плохой прогноз и тяжелые неврологические нарушения, обычно с ранней смертью».
Управление
[ редактировать ]Ключевой проблемой является раннее сращение черепа, которое можно исправить с помощью ряда хирургических вмешательств, часто в течение первых трех месяцев после рождения. Более поздние операции необходимы для коррекции респираторных и лицевых деформаций. [2]
Результаты
[ редактировать ]Дети с синдромом Пфайффера 2 и 3 типа «имеют более высокий риск нарушений нервного развития и меньшую продолжительность жизни», чем дети с синдромом Пфайффера 1 типа, но при лечении возможны благоприятные исходы. [14] В тяжелых случаях респираторные и неврологические осложнения часто приводят к ранней смерти.
История
[ редактировать ]Синдром назван в честь немецкого генетика Рудольфа Артура Пфайффера (1931–2012). [15] В 1964 году Пфайффер описал восемь человек в трех поколениях семьи, у которых были аномалии головы, рук и ног ( акроцефалосиндактилия ), наследуемые по аутосомно-доминантному типу. [1] [13] [3]
Известные случаи
[ редактировать ]- родился сын В 1996 году у американского музыканта Принса и его жены Майте Гарсиа . У долгожданного ребенка, Амира («принца» по-арабски), при рождении был диагностирован синдром Пфайффера 2 типа, и он умер через несколько дней. [16] В 1997 году, после того как бывшие личные помощники Гарсии выразили обеспокоенность по поводу способа смерти, судмедэксперт провел расследование и заявил, что смерть наступила по естественным причинам (то есть это не было убийством). [17]
- В 2014 году мать мальчика из Техаса с синдромом Пфайффера 1 типа разместила в своем блоге фотографию ребенка. В 2016 году она обнаружила, что фотография была использована в меме, сравнивающем ее сына с мопсом . Ее усилия по удалению мема из Интернета, особенно из социальных сетей, таких как Instagram , Twitter и Facebook , привлекли международное внимание. [18] [19] [20]
Ссылки
[ редактировать ]- ^ Jump up to: а б с д и ж г Фогельс А., Фринс Дж. П. (2006). «Синдром Пфайффера» . Orphanet J Редкий дис . 1:19 . дои : 10.1186/1750-1172-1-19 . ПМЦ 1482682 . ПМИД 16740155 .
- ^ Jump up to: а б с «Синдром Пфайфера» . НОРД (Национальная организация по редким заболеваниям) . Проверено 03 сентября 2019 г.
- ^ Jump up to: а б Пфайффер Р.А. (1964). «Доминантная наследственная акроцефалосиндактилия». Журнал педиатрии (на немецком языке). 90 (4): 301–20. дои : 10.1007/BF00447500 . ПМИД 14316612 . S2CID 35706808 .
- ^ «Синдром Пфайффера» . Национальная медицинская библиотека США . Проверено 29 октября 2020 г.
- ^ «Детский синдром Пфайфера» . Детский национальный . Проверено 24 февраля 2023 г.
- ^ «Синдром Пфайффера» . Национальная медицинская библиотека США . Проверено 29 октября 2020 г.
- ^ Муенке М; Шелл У; Гер А; Робин Н.Х.; Лоскен Х.В.; Шинцель А; и др. (1994). «Распространенная мутация в гене рецептора 1 фактора роста фибробластов при синдроме Пфайффера». Нат Жене . 8 (3): 269–74. дои : 10.1038/ng1194-269 . ПМИД 7874169 . S2CID 40033932 .
- ^ Ратленд П; Пуллейн Л.Дж.; Рирдон В; Барайцер М; Хейворд Р; Джонс Б; и др. (1995). «Идентичные мутации в гене FGFR2 вызывают фенотипы синдрома Пфайффера и Крузона». Нат Жене . 9 (2): 173–6. дои : 10.1038/ng0295-173 . ПМИД 7719345 . S2CID 927144 .
- ^ Шелл У., Хер А., Фельдман Г.Дж., Робин Н.Х., Закай Э.Х., де Ди-Смолдерс С. и др. (1995). «Мутации в FGFR1 и FGFR2 вызывают семейный и спорадический синдром Пфайффера». Хум Мол Жене . 4 (3): 323–8. дои : 10.1093/hmg/4.3.323 . ПМИД 7795583 .
- ^ Чан CT, Торогуд П. (1999). «Плейотропные особенности синдромальных краниосиностозов коррелируют с дифференциальной экспрессией рецепторов фактора роста фибробластов 1 и 2 во время черепно-лицевого развития человека» . Педиатр. Рез . 45 (1): 46–53. дои : 10.1203/00006450-199901000-00008 . ПМИД 9890607 .
- ^ Глейзер Р.Л., Цзян В., Бояджиев С.А., Тран А.К., Закари А.А., Ван Малдергем Л. и др. (2000). «Отцовское происхождение мутаций FGFR2 в спорадических случаях синдрома Крузона и синдрома Пфайффера» . Ам Джей Хум Жене . 66 (3): 768–77. дои : 10.1086/302831 . ПМК 1288162 . ПМИД 10712195 .
- ^ Информационный центр Национального института здравоохранения, генетических и редких заболеваний (GARD) (01 апреля 2016 г.). «Синдром Пфайффера: Симптомы» . Проверено 8 мая 2016 г.
- ^ Jump up to: а б Коэн ММ (1993). «Обновленные данные о синдроме Пфайффера, клинические подтипы и рекомендации по дифференциальной диагностике». Am J Med Genet . 45 (3): 300–7. дои : 10.1002/ajmg.1320450305 . ПМИД 8434615 .
- ^ Робин Н.Х.; Скотт Дж.А.; Арнольд Дж.Э.; Гольдштейн Дж.А.; Шиллинг ББ; Мэрион Р.В.; и др. (1998). «Благоприятный прогноз для детей с синдромом Пфайффера 2 и 3 типа: значение для классификации». Am J Med Genet . 75 (3): 240–4. doi : 10.1002/(sici)1096-8628(19980123)75:3<240::aid-ajmg2>3.3.co;2-c . ПМИД 9475589 .
- ^ Synd/3477 в Who Named It?
- ^ Лернер, Маура (28 марта 1997 г.). «Судебное дело Принса затрагивает два вопроса: право семьи на неприкосновенность частной жизни и медицинскую этику». Звездная Трибьюн . Миннеаполис – через NewsBank.
- ^ Чанен, Дэвид (14 июня 1997 г.). «Решение: ребенок Принса умер естественной смертью». Звездная Трибьюн . Миннеаполис – через NewsBank.
- ^ Пеллетьери, Николь (2 февраля 2016 г.). «Мама защищает сына от оскорбительного интернет-мема» . Новости АВС . Проверено 8 мая 2016 г.
- ^ Эрнандес, Витторио (5 февраля 2016 г.). «Мама из Техаса разозлилась из-за использования фотографии сына с редким расстройством синдрома Пфайффера для создания жестоких мемов» . International Business Times , австралийское издание . Проверено 8 мая 2016 г.
- ^ «Мать защищается от шуток в адрес ее больного сына » . Штерн (на немецком языке). 08 февраля 2016 г. Проверено 8 мая 2016 г.
Внешние ссылки
[ редактировать ]- Робин, Нью-Хэмпшир; Фальк, MJ; Халдеман-Энглерт, CR (07.06.2011) [первоначальная публикация 1998 г.]. «Обзор синдромов краниосиностоза FGFR» . Синдромы краниосиностоза, связанные с FGFR . Джин Обзоры . НКБИ . ПМИД 20301628 .
- Хамм, А; Робин, Н. (октябрь 2014 г.). «Синдром Пфайфера» . Сирота . МКБ-10 Q87.0 .