Acrocephalosyndactyly
| Acrocephalosyndactyly | |
|---|---|
| Другие имена | Кондиционер |
 | |
| Синдактилия в acrocephalosyndactyly (apert) | |
| Специальность | Медицинская генетика |
Acrocephalosyndactyly - это группа врожденных состояний, характеризующихся нерегулярными особенностями лица и черепа ( краниосиностоз ), и руками и ногами ( синдактилия ). [ 1 ] Краниосиностоз возникает, когда черепные швы, волокнистая ткань, соединяющая кости черепа, слияет черепные кости на ранних этапах развития. Черновые швы позволяют костям черепа продолжать расти до тех пор, пока они не будут сплачиваться в возрасте 24 лет. Преждевременное слияние черепных швов может привести к изменениям формы черепа и мешать росту мозга. [ 2 ] [ 3 ] Синдактилия происходит, когда цифры рук или ног сливаются вместе. [ 4 ] Когда также присутствует полидактилия , классификация является акроцефалополизиндактической. [ 5 ] Полидактилия происходит, когда руки или ноги обладают дополнительными цифрами. [ 6 ] Акроцефалосиндактилия обычно диагностируется после рождения, хотя пренатальная диагностика иногда возможна, если генетическая вариация присутствует у членов семьи, поскольку условия обычно наследуют в аутосомно -доминантном схеме [ 1 ] Лечение часто включает в себя операцию в раннем детстве, чтобы исправить краниосиностоз [ 7 ] и синдактилия . [ 8 ]
Тяжесть симптомов для акроцефалосиндактилии значительно варьируется в зависимости от подтипа и лечения на ранних стадиях жизни.
История
[ редактировать ]Случаи состояния были зарегистрированы еще в 18 веке. [ 9 ] Термин Acrocephalosyndactyly (от греческого ἄκρος ( ákros) 'самая высокая, на конечности », κεφαλή (kephalḗ) ' голова ', σύν (syn) ' вместе 'и Δάκτυλος (daktylos) ' finger ') был сначала применен во французском физическом. Eugène Apert сначала для описания состояния, характеризующегося краниосиностозом и синдилия . [ 10 ] Состояние, описанное Apert, в настоящее время известно как подтип синдрома аперта акроцефалосиндактилия. [ 11 ] Другие подтипы акроцефалосиндактилии были охарактеризованы в течение 20 -го века. [ 1 ]
Распространенность
[ редактировать ]Учитывая все виды акроцефалосиндактилии, один новорожденный ребенок рождается с акроцефалосиндактами на каждые 65 000 - 102 500 детей. Не существует разницы в количестве мужчин и женщин, затронутых акроцефалосиндактилия. [ 12 ] [ 13 ] [ 14 ] [ 15 ]
Характеристики
[ редактировать ]Акроцефалосиндактилия представляет во многих различных подтипах, однако происходит значительное совпадение в симптомах. Как правило, все формы акроцефалосиндактилии характеризуются нетипичными черепно -лицевыми, ручными и ногами, такими как преждевременное закрытие фиброзных суставов между определенными костями черепа, [ 16 ] [ 17 ] слияние определенных рук или ног, [ 16 ] [ 18 ] и/или больше, чем обычное количество цифр. [ 19 ] Некоторые подтипы также включают структурные изменения сердца , которые присутствуют при рождении. [ 20 ] [ 1 ]
Причина
[ редактировать ]Большинство форм акроцефалосиндактилии или акроцефалополизиндактилии наследуются в аутосомно -доминантной картине, [ 15 ] [ 20 ] [ 1 ] Благодаря исключению синдрома Карпентера , который унаследован аутосомно -рецессивным образом. De-Novo Варианты , или генетические изменения, не унаследованные от родителей, сообщалось, что в разных генах вызывают несколько типов акроцефалосиндактилии. Среди известных генетических изменений существуют различия в таких генах, как рецептор фактора роста фибробластов ( FGFR ) , [ 16 ] [ 17 ] [ 21 ] [ 22 ] Twist1 , [ 23 ] и Rab23 . [ 24 ] Конститутивная активация в этих категориях генов, в частности FGF R, которая активно участвует на стадии развития эмбрионов, таких как развитие органов или органогенез , и поддержание тканевых клеток, известных как клетки -предшественники , может быть очень вредной. [ 25 ]
Генетически унаследованные акроцефалосиндактильные условия - все они показывают высокую, чтобы завершить проникновение с переменной экспрессией , что означает, что все люди, которые наследуют состояние, присутствующее нетипичное характеристическое черепно -лицевое, ручное и стоп -структуры, но тяжесть инвалидности является переменной. [ 1 ] Повышение отцовского возраста считается фактором риска в некоторых случаях. [ 1 ]
Воздействие условий на жизнь
[ редактировать ]Несмотря на нынешние серьезные усилия хирургической терапии на влияние акроцефалосиндактилии, заболевания все еще существуют у людей, которые получали лечение. Те, кто достигает взрослой жизни, часто имеют более низкий уровень образования, чем их сверстники, а также большие трудности в различных социальных аспектах, таких как свидания, брак или сексуальные отношения. Они также могут сообщить о необходимости жизни в жизни на протяжении всей своей жизни, а также о других проблемах со здоровьем, таких как проблемы слуха или эпилепсия на более распространенной частоте, чем их коллеги. [ 26 ]
К счастью, многие люди с этим условием сообщают о сходных уровнях счастья со своей жизнью как без аффиль [ 26 ] и показать высокую социальную интеграцию , а также большую физическую и эмоциональную устойчивость, несмотря на любые препятствия. [ 27 ]
Диагноз
[ редактировать ]Пренатальный диагноз
[ редактировать ]Диагноз до рождения возможен для некоторых форм акроцефалосиндактилии. Пренатальная генетическая диагностика возможен только в том случае, если известно изменение гена, ответственное за синдром, и вариация, вызывающая заболевание, было идентифицировано в геноме члена семьи. Сбор образцов для генетического тестирования может быть выполнена с использованием амниоцентеза , который образцы эмбриональных стволовых клеток, содержащихся в амниотической жидкости, или отбора проб хорионической ворсинки , которые обрабатывают плацентарные клетки. [ 15 ] Был случай пренатального диагноза синдрома Аперта с использованием фетоскопии , где плод наблюдается с использованием эндоскопа, вставленного в матку из живота. [ 22 ] Альтернативно, был заинтересован в использовании неинвазивных методов, таких как ультразвук для обнаружения нетипичных особенностей черепа плода. [ 28 ]
Постнатальный диагноз
[ редактировать ]Большинство диагнозов акроцефалосиндактилии возникают после рождения путем оценки физических симптомов младенца. Это может быть подтверждено рентгенографической визуализацией, такой как рентгеновская визуализация и молекулярное генетическое тестирование , которое ищет вариации ДНК, которые, как известно, вызывают заболевание. [ 29 ] [ 30 ] Молекулярное генетическое тестирование обычно происходит в генах FGFR , Twist1 и Rab23 .
Номенклатура/классификация
[ редактировать ]Не существует последовательной номенклатуры или классификации в различных синдромах под зонтиком акроцефалосиндактации и акроцефалополизиндактации. Хотя акроцефалосиндактилия сообщалось еще в 18 веке, [ 9 ] Классификации ACS и ACPS появились только в последнем 20 -м веке. Тем не менее, эта классификация может быть устарела, поскольку было предположено, что различие между акроцефалосиндактами и акроцефалополизиндактацией должно быть стерто. [ 5 ]
В настоящее время синдром Нока (ACPS тип I) теперь классифицируется как синдром Пфайффера (тип V ACS V); [ 31 ] Синдром Гудмана (ACPS тип IV) классифицируется как вариация синдрома Карпентера (тип ACPS II); [ 19 ] и разные исследователи объединили APERT (тип ASC I), Crouzon (ASC Type II) и Pfeiffer (ASC Type V) в аперт-кроузон синдром [ 32 ] и Crouzon-Pfeiffer [ 33 ] синдром.
Акроцефалосиндактилия типа IV ранее назывался синдромом MOHR, однако позже он был классифицирован под синдромом орофациодигита, тип II. [ 34 ] Синдром Пфайффера ранее был типа VI и Ваарденбурга типа V, но это изменилось через некоторое время после 1966 года. [ 35 ]
Acrocephalosyndactyly (acs):
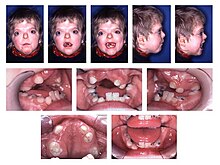
- Тип I - Синдром Аперта [ 11 ]
- Тип II - синдром Крузона [ 36 ]
- Тип III - Синдром Saethre -Chotzen [ 37 ]
- Синдром Робинов-Сорауф предполагается, что в классификации Saethre-Chotzen Classification [ 38 ]
- Тип IV - синдром MOHR (архаика) [ 34 ] [ 35 ]
- Тип V - синдром Пфайффера [ 31 ]
- Синдром Нока включен в классификацию синдрома Пфайффера [ 31 ]
Связанный термин, акроцефалополизиндактилия (ACP), относится к включению полидактилии к презентации. Он также имеет несколько типов:
- Тип I - Синдром Нока (архаичный) [ 31 ]
- Тип II - синдром плотника [ 39 ]
- Синдром Гудмана включен в синдрома Карпентера классификацию [ 40 ]
- Синдром Саммитта включен в синдрома Карпентера классификацию [ 41 ]
- Тип II- Синдром типа II- Сакати-Нихан-Тисдейл [ 42 ]
- Тип IV - синдром Гудмана (архаичный) [ 40 ]
Уход
[ редактировать ]Краниосиностоз
[ редактировать ]Для подтипов с краниосиностозом необходима хирургия для предотвращения преждевременного слияния черепных швов, таких как корональный швар ( брахицефалия ). [ 7 ] Черновой шов, расположенный между двумя фронтальными и двумя теменными костями черепа, называется корональным швом. [ 2 ] Краниопластика должна быть выполнена в первый год жизни, чтобы предотвратить нарушения роста мозга из -за повышенного внутричерепного давления. Хирургия среднего звена может также потребоваться в детстве, чтобы отделить среднее место от остальной части черепа для исправления респираторных и ортодонтических проблем. [ 43 ] [ 44 ]
Синдактилия
[ редактировать ]Синдактилия в определенных подтипах редко бывает достаточно тяжелой, чтобы повлиять на функцию рук, поэтому лечение может не понадобиться. [ 43 ]
В более серьезных подтипах, как видно при синдроме Аперта , может потребоваться хирургическая коррекция синдилией. Хирургия рекомендуется выполнять как можно скорее, как правило, в возрасте 4 месяцев. Лечение зависит от тяжести синдизии. Хирургическое лечение, как правило, включает в себя междигитальный релиз веб -пространства и удлинение большого пальца. [ 8 ]
Управление
[ редактировать ]Лечение для тех, у кого диагностирован акроцефалосиндактилия, простирается за пределы операции. Есть много шагов, которые могут помочь в долгосрочном управлении синдромом. Люди, страдающие от акроцефалосиндактилии, и их лиц, осуществляющие уход, могут построить систему поддержки здравоохранения, построив прочные отношения с командой медицинских специалистов. Предварительно сформированные команды медицинских специалистов часто можно найти в университетах или в исследовательских учреждениях . Уход за больными могут предотвратить будущие проблемы, изучая варианты финансовой помощи , медицинского страхования и предоставления учебных заведений. Основным лицам, опечивающим, рекомендуется расставлять приоритеты в своем эмоциональном здоровье, оставляя время для себя и поставляя надежную систему поддержки. [ 29 ]
Смотрите также
[ редактировать ]Ссылки
[ редактировать ]- ^ Jump up to: а беременный в дюймовый и фон глин Биссоннетт, Бруно; Luginbuehl, Igor; Marciniak, Bruno; Dalens, Bernard J. (2006), «Акроцефалосиндактильные синдромы» , Синдромы: быстрое распознавание и периоперационные последствия , Нью-Йорк, Нью-Йорк: компании McGraw-Hill Companies
- ^ Jump up to: а беременный Рассел, Уильям П.; Рассел, Марк Р. (2023), «Анатомия, голова и шея, корональный шар» , Statpearls , Island Treasure (FL): Patpearls Publishing, PMID 30252267 , извлеченные 4 декабря 2023 г.
- ^ Opperman, LA (2000). «Черновые швы как внутримембрановые участки роста костей» . Динамика развития . 219 (4): 472–485. doi : 10.1002/1097-0177 (2000) 9999: 9999 <:: apid-dvdy1073> 3.0.co; 2-f . ISSN 1058-8388 . PMID 11084647 . S2CID 8801611 .
- ^ Малик С. (2012). «Синдактилия: фенотипы, генетика и текущая классификация» . Европейский журнал человеческой генетики . 20 (8): 817–824. doi : 10.1038/ejhg.2012.14 . ISSN 1476-5438 . PMC 3400728 . PMID 22333904 .
- ^ Jump up to: а беременный Коэн, М. Майкл; Крейборг, Свен (май 1995). «Руки и ноги в синдроме Аперта» . Американский журнал медицинской генетики . 57 (1): 82–96. doi : 10.1002/ajmg.1320570119 . ISSN 0148-7299 . PMID 7645606 .
- ^ Малик С. (2014). «Полидактилия: фенотипы, генетика и классификация» . Клиническая генетика . 85 (3): 203–212. doi : 10.1111/cge.12276 . ISSN 0009-9163 . PMID 24020795 . S2CID 22412404 .
- ^ Jump up to: а беременный Краниосиностоз: диагностика, оценка и лечение . М. Майкл Коэн, Рут Э. Маклин (2 -е изд.). Нью -Йорк: издательство Оксфордского университета. 2000. ISBN 0-19-511843-X Полем OCLC 41528658 .
{{cite book}}: Cs1 maint: другие ( ссылка ) - ^ Jump up to: а беременный Рапосо-амарал, Кассио Эдуардо; Денадай, Рафаэль; Фурлан, Педро; Рапосо-амарал, Сезар Аугусто (Octber 2018). «Лечение синдрома Apert Hand: стратегии достижения пятизначной руки» . Пластическая и реконструктивная хирургия . 142 (4): 972–982. Doi : 10.1097/prs.0000000000004815 . ISSN 0032-1052 . PMID 29994846 . S2CID 51614940 .
- ^ Jump up to: а беременный Уитон, SW (1894). «Два образца врожденной граниальной деформации у детей, связанных с слиянием пальцев рук и ног». Trans Pathol Soc Lond . 45 : 238–241.
- ^ Аперт, я (1906). "De l'crocephalosyndactylie". Бык Мем. Соц Méd. Прыгать. Париж . 23 : 1310–1330.
- ^ Jump up to: а беременный «Вход - #101200 - Синдром Аперта» . Omim.org . Получено 4 декабря 2023 года .
- ^ М Дас, Джо; Winters, Райан (2023), «Синдром Пфайффера» , Statpearls , Island Treasure (FL): издательство Statpearls, PMID 30422477 , получено 4 декабря 2023 г.
- ^ Конради, Кристофер Д.; Патель, Бхупендра С.; Шарма, Сандип (2023), «Синдром Аперта» , Statpearls , Island Treasure (FL): Statpearls Publishing, PMID 30085535 , получен 4 декабря 2023 г.
- ^ Tolorova, MM (2023). «Генетика синдрома Крузона» . Педиатрия: генетика и метаболическое заболевание - через Medscape.
- ^ Jump up to: а беременный в Галлахер, Эмили Р.; Ratisoontorn, chootima; Каннингем, Майкл Л. (1993), Адам, Маргарет П.; Фельдман, Джерри; Mirzaa, Ghayda M.; Pagon, Roberta A. (Eds.), «Синдром Сэтре-Чотцен» , Genereviews® , Seattle (WA): Университет Вашингтона, Сиэтл, PMID 20301368 , получен 4 декабря 2023 года.
- ^ Jump up to: а беременный в Уилки, Эндрю Ом; Слни, Сара Ф.; Олдридж, Майкл; Пул, Майкл Д.; Эшворт, Джеральдин Дж.; Хокли, Энтони Д.; Хейворд, Ричард Д.; Дэвид, Дэвид Дж.; Пуллин, Луиза Дж.; Ратленд, Пол; Малкольм, Сьюзен; Зима, Робин М.; Рирдон, Уильям (февраль 1995 г.). «Синдром Аперта является результатом локализованных мутаций FGFR2 и является аллельным синдромом Крузона» . Природа генетика . 9 (2): 165–172. doi : 10.1038/ng0295-165 . ISSN 1546-1718 . PMID 7719344 . S2CID 12423131 .
- ^ Jump up to: а беременный Каринси, Франческо; Части, Фурио; Locci, Paola; Бекчетти, Эннио; Карлс, Фридрик; Avantaggiato, Anna; Бекчетти, Алессио; Каринси, Паоло; Барони, Тизиано; Бодо, Мария (май 2005 г.). «Синдромы Аперта и Крузона: клинические результаты, гены и внеклеточный матрикс» . Журнал черепно -лицевой хирургии . 16 (3): 361–368. Doi : 10.1097/01.scs.0000157078.53871.11 . ISSN 1049-2275 . PMID 15915098 . S2CID 23327865 .
- ^ Биссоннетт, Бруно; Luginbuehl, Igor; Engelhardt, Thomas (2019), «Акроцефалополизиндактилия типа IV: Синдром Гудмана» , Синдромы: быстрое признание и периоперационные последствия (2 изд.), Нью-Йорк, Нью-Йорк: McGraw-Hill Education
- ^ Jump up to: а беременный Коэн, Дональд М.; Грин, Джеймс Г.; Миллер, Дженис; Горлин, Роберт Дж.; Рид, Джерри А.; Opitz, John M.; Рейнольдс, Джеймс Ф. (октябрь 1987 г.). «Акроцефалополизиндактилия типа II - Синдром Карпентера: клинический спектр и попытка объединения с синдромами Гудмана и Саммита» . Американский журнал медицинской генетики . 28 (2): 311–324. doi : 10.1002/ajmg.1320280208 . ISSN 0148-7299 . PMID 3322002 .
- ^ Jump up to: а беременный Кумар, Нирадж; Арора, Шубханги; Биндра, Ашиш; Гоял, Кешав (2014). «Анестезиозное лечение восстановления краниосиностоза у пациента с синдромом Аперта» . Саудовский журнал анестезии . 8 (3): 399–401. doi : 10.4103/1658-354x.136631 . ISSN 1658-354x . PMC 4141395 . PMID 25191197 .
- ^ Галвин, BD; Харт, KC; Мейер, Ан; Вебстер, MK; Donoghue, DJ (23 июля 1996 г.). «Конститутивная активация рецепторов мутациями синдрома Крузона в рецепторе фактора роста фибробластов (FGFR) 2 и FGFR2/Neu Chimeras» . Труды Национальной академии наук . 93 (15): 7894–7899. Bibcode : 1996pnas ... 93,7894G . doi : 10.1073/pnas.93.15.7894 . ISSN 0027-8424 . PMC 38845 . PMID 8755573 .
- ^ Jump up to: а беременный Леонард, Клэр О.; Дайкоку, Норман Х.; Винн, Кевин; Опиц, Джон М. (январь 1982). «Пренатальная фетоскопическая диагностика синдрома аперта» . Американский журнал медицинской генетики . 11 (1): 5–9. doi : 10.1002/ajmg.1320110103 . ISSN 0148-7299 . PMID 7065003 .
- ^ Cai, Juanliang; Гудман, Барбара К.; Патель, Анкита С.; Малликен, Джон Б.; Ван Малдерж, Лайонел; Хогансон, Джордж Э.; Пацнекас, Уильям А.; Бен-Мерия, Зива; Шеффер, Рут; Каннингем, Майкл Л.; Daentl, Donna L.; Джабс, этилин Ван (1 декабря 2003 г.). «Повышенный риск задержки развития при синдроме Сэтре-Чотзена связан с удалением поворота: улучшенной стратегией скрининга мутаций Twist» . Человеческая генетика . 114 (1): 68–76. doi : 10.1007/s00439-003-1012-7 . ISSN 1432-1203 . PMID 14513358 . S2CID 20929600 .
- ^ Дженкинс, Даган; Силоу, Доминик; Джехи, Фернанда С.; Перлин, Чад А.; Алонсо, Луис Г.; Буэно, Даниэла Ф.; Доннаи, Диан; Джозифиова, Драгана; Mathijssen, Irene MJ; Мортон, Дженни Э.В.; Эрставик, Карен Хелен; Суини, Элизабет; Стена, Стивен А.; Марш, Джеффри Л.; Нюрнберг, Питер (1 июня 2007 г.). «Мутации Rab23 при синдроме Карпентера подразумевают неожиданную роль передачи сигналов ежа при развитии и ожирении черепной и ожирения» . Американский журнал человеческой генетики . 80 (6): 1162–1170. doi : 10.1086/518047 . ISSN 0002-9297 . PMC 1867103 . PMID 17503333 .
- ^ Ямаджи, Кодзиро ; синдрома Аперта " . Динамика развития . 247 (11): 1175–1185. DOI : /DVDDY.24673 . ISSN 1058-8388 . PMID 30251381. S2CID 10.1002 52815441 .
- ^ Jump up to: а беременный Tovetjärn, Роберт; Тарнов, Петр; Мальтийский, Джованни; Фишер, Сара; Sahlin, Per-erik; Кёльби, Ларс (октябрь 2012 г.). «Дети с синдромом Аперта в качестве взрослых: последующее исследование 28 скандинавских пациентов» . Пластическая и реконструктивная хирургия . 130 (4): 572e - 576e. doi : 10.1097/prs.0b013e318262f355 . ISSN 0032-1052 . PMID 23018718 . S2CID 45847015 .
- ^ Тагиния, Амир Х.; Yorlets, Rachel R.; Дойл, Майкл; Лабоу, Брайан I.; Аптон, Джозеф (апрель 2019 г.). «Долгосрочные функциональные результаты верхней значимости у взрослых с синдромом Аперта» . Пластическая и реконструктивная хирургия . 143 (4): 1136–1145. doi : 10.1097/prs.0000000000005479 . ISSN 0032-1052 . PMID 30676503 . S2CID 59225959 .
- ^ «Энциклопедия генетических расстройств и врожденных дефектов | WorldCat.org» . search.worldcat.org . Получено 4 декабря 2023 года .
- ^ Jump up to: а беременный «Синдром Аперта - Диагностика и лечение - Информационный центр генетических и редких заболеваний» . rarediseases.info.nih.gov . Получено 5 ноября 2022 года .
- ^ Gonzales, M.; Heuertz, S.; Martinovic, J.; Delahaye, S.; Базин, А.; Loget, P.; Pasquier, L.; Le Merrer, M.; Bonaventure, J. (17 июня 2005 г.). «Аномалии позвонков и хрящевая рукав трахеи у трех пациентов с синдромом Пфайффера, несущей мутацию S351C FGFR2: буква к редактору» . Клиническая генетика . 68 (2): 179–181. doi : 10.1111/j.1399-0004.2005.00477.x . PMID 15996217 . S2CID 1652216 .
- ^ Jump up to: а беременный в дюймовый «Вход № 101600 - Синдром Пфайффера» . Omim.org . Получено 4 декабря 2023 года .
- ^ Rédei, George P., ed. (2008), «Синдром Аперта или Аперт-Крузон» , Энциклопедия генетики, геномики, протеомики и информатики , Дордрехт: Спрингер Нидерланды, с. 127, doi : 10.1007/978-1-4020-6754-9_1009 , ISBN 978-1-4020-6754-9 , Получено 5 ноября 2022 года
- ^ Уилсон, Александр Т.; De Planque, Екатерина А.; Ян, Сумин С.; Tasker, Robert C.; Van Veelen, Marie-Lise C.; Dremmen, Marjolein Hg; Vrooman, Anri A.; Матийсен, Ирен М.Дж. (октябрь 2020 г.). «Толщина коры при синдроме Крузон-Пфайффера: результаты в отношении первичного расширения хранилища черепа» . Пластическая и реконструктивная хирургия. Global Open . 8 (10): E3204. doi : 10.1097/gox.0000000000003204 . ISSN 2169-7574 . PMC 7647527 . PMID 33173703 .
- ^ Jump up to: а беременный «Вход - %252100 - Синдром Мор» . Omim.org . Получено 5 ноября 2022 года .
- ^ Jump up to: а беременный McKusick, VA (1966). «Аутосомно -доминантные фенотипы». Менделянское наследство у человека: каталоги аутосомно-доминантных, аутосомно-рецессивных и x-связанных фенотипов . Johns Hopkins Press. С. 3–5.
- ^ «Вход - #123500 - Синдром Крузона - Омим» . Omim.org . Получено 5 ноября 2022 года .
- ^ «Вход - #101400 - Синдром Сэтре -Чотцен; SCS - Omim» . Omim.org . Получено 5 ноября 2022 года .
- ^ REARDON, W.; Зима, RM (1994). «Синдром Сэтре-Чотзена» . Журнал медицинской генетики . 31 (5): 393–396. doi : 10.1136/jmg.31.5.393 . ISSN 0022-2593 . PMC 1049872 . PMID 8064818 .
- ^ «Вход № 201000 - Синдром Карпентера 1; CRPT1» . Omim.org . Получено 4 декабря 2023 года .
- ^ Jump up to: а беременный «Вход 201020 - Acrocephalopolysyndactyly Type IV» . Omim.org . Получено 4 декабря 2023 года .
- ^ «Вход 272350 - Синдром Саммитта» . Omim.org . Получено 4 декабря 2023 года .
- ^ «Вход 101120 - Acrocepholopolysyndactyly Type III» . Omim.org . Получено 4 декабря 2023 года .
- ^ Jump up to: а беременный Андерсон, Питер. «Заголовки черепно -лицевой поддержки» (PDF) . Архивировано из оригинала 30 июня 2012 года.
- ^ «Синдром Пфайффера» . Норд (Национальная организация редких расстройств) . Получено 5 ноября 2022 года .
Внешние ссылки
[ редактировать ]- Acrocephalosyndactylia в Национальной медицинской библиотеке Медицинской библиотеки США (Mesh)