синдром Кокейна
| синдром Кокейна | |
|---|---|
| Другие имена | Синдром Нила-Дингволла |
 | |
| Специальность | Медицинская генетика , неврология , дерматология |
Синдром Кокейна ( CS ), также называемый синдромом Нила-Дингволла , представляет собой редкое и смертельное аутосомно- рецессивное нейродегенеративное заболевание, характеризующееся задержкой роста, нарушением развития нервной системы , аномальной чувствительностью к солнечному свету ( светочувствительностью ), нарушениями зрения и преждевременным старением . [1] [2] [3] отставание в развитии и неврологические расстройства, а другими очень распространенными признаками являются светочувствительность, потеря слуха, аномалии глаз и кариес. Критериями диагноза являются [3] Возможны проблемы с любым или всеми внутренними органами. Это связано с группой расстройств, называемых лейкодистрофиями , которые представляют собой состояния, характеризующиеся деградацией неврологического белого вещества . Существует два основных типа синдрома Коккейна: синдром Коккейна типа А (CSA) , возникающий в результате мутаций гена ERCC8 , и синдром Коккейна типа B (CSB) , возникающий в результате мутаций гена ERCC6 . [4]
В основе заболевания лежит дефект механизма репарации ДНК . [5] В отличие от других дефектов репарации ДНК, пациенты с КС не предрасположены к раку или инфекциям. [6] Синдром Кокейна — редкое, но разрушительное заболевание, обычно приводящее к смерти в течение первого или второго десятилетия жизни. Мутация специфических генов при синдроме Коккейна известна, но широко распространенные последствия и ее связь с репарацией ДНК еще предстоит хорошо понять. [6]
Он назван в честь английского врача Эдварда Альфреда Кокейна (1880–1956), который впервые описал его в 1936 году и повторно описал в 1946 году. [7] Синдром Нила-Дингволла был назван в честь Мэри М. Дингуолл и Кэтрин А. Нил. [7] Эти два учёных описали случай двух братьев с синдромом Кокейна и утверждали, что это то же самое заболевание, которое описал Кокейн. В своей статье они внесли свой вклад в появление признаков заболевания, обнаружив кальцификаты в мозге. Они также сравнили синдром Кокейна с тем, что сейчас известно как синдром прогерии Хатчинсона-Гилфорда (HGPS), который тогда назывался прогерией, из-за преждевременного старения, которое характеризует оба расстройства. [7]
Типы
[ редактировать ]- КС I типа, «классическая» форма, характеризуется нормальным развитием плода с появлением отклонений в первые два года жизни. Зрение и слух постепенно ухудшаются. [8] Центральная и периферическая нервные системы прогрессивно дегенерируют вплоть до смерти в первом или втором десятилетии жизни в результате серьезной неврологической деградации. Кортикальная атрофия менее выражена при CS I типа. [9]
- CS типа II присутствует с рождения ( врожденный ) и протекает гораздо тяжелее, чем CS типа 1. [8] После рождения он включает в себя очень незначительное неврологическое развитие. Смерть обычно наступает к семи годам. Этот конкретный тип также был обозначен как церебро-окуло-фацио-скелетный синдром (COFS) или синдром Пена-Шокейра типа II. [8] Синдром COFS назван так из-за воздействия, которое он оказывает на мозг, глаза, лицо и скелет, поскольку заболевание часто вызывает атрофию мозга, катаракту, потерю жира на лице и остеопороз. Синдром COFS можно подразделить на несколько состояний (типы COFS 1, 2, 3 (связанные с пигментной ксеродермией ) и 4). [10] Обычно у пациентов с этой формой заболевания с ранним началом наблюдаются более серьезные повреждения головного мозга, включая снижение миелинизации белого вещества и более распространенную кальцификацию, в том числе в коре головного мозга и базальных ганглиях. [9]
- CS типа III, характеризующегося поздним началом, обычно протекает мягче, чем типы I и II. [8] Часто пациенты с типом III доживают до взрослого возраста.
- Пигментная ксеродермия-синдром Коккейна (XP-CS) возникает, когда у человека также имеется пигментная ксеродермия, еще одно заболевание, связанное с репарацией ДНК. Выражены некоторые симптомы каждого заболевания. Например, присутствуют веснушки и пигментные нарушения, характерные для XP. Отмечаются неврологические расстройства, спастичность и недоразвитие половых органов, характерные для КС. Однако гипомиелинизация и черты лица типичных пациентов с КС отсутствуют. [11]
Причины
[ редактировать ]Если гипероксия или избыток кислорода в организме возникает , клеточный метаболизм производит несколько высокореактивных форм кислорода, называемых свободными радикалами . Это может вызвать окислительное повреждение клеточных компонентов, включая ДНК . В нормальных клетках наш организм восстанавливает поврежденные участки. В случае этого заболевания из-за незначительных дефектов транскрипции детский генетический механизм синтеза белков, необходимых организму, не работает в нормальном режиме. Согласно этой теории, со временем это приводит к задержке развития и смерти. Каждую минуту организм перекачивает через кровь от 10 до 20 литров кислорода , доставляя его к миллиардам клеток нашего тела. В своей нормальной молекулярной форме кислород безвреден. Однако клеточный метаболизм с участием кислорода может генерировать несколько высокореактивных свободных радикалов. Эти свободные радикалы могут вызвать окислительное повреждение клеточных компонентов, включая ДНК. В средней человеческой клетке несколько тысяч повреждений каждый день в ДНК происходит . Многие из этих поражений являются результатом окислительного повреждения. . Каждое повреждение — поврежденный участок ДНК — должно быть вырезано, а ДНК восстановлена, чтобы сохранить свою нормальную функцию. Невосстановленная ДНК может потерять способность кодировать белки. Также могут возникнуть мутации. Эти мутации могут активировать онкогены или заглушить гены-супрессоры опухолей. Согласно исследованиям, окислительное повреждение активных генов преимущественно не восстанавливается, а в наиболее тяжелых случаях восстановление замедляется по всему геному . Возникающее в результате накопление окислительного повреждения может нарушить нормальные функции ДНК и даже привести к запуску программы гибели клеток (апоптоза). Дети с этим заболеванием не восстанавливают активные гены, вызывающие окислительное повреждение. Обычно восстановление окислительных повреждений происходит быстрее в активных генах (которые составляют менее пяти процентов генома), чем в неактивных участках ДНК. Возникающее в результате накопление окислительного повреждения может нарушить нормальные функции ДНК и даже привести к запуску программы гибели клеток. апоптоз ). [12]
Генетика
[ редактировать ]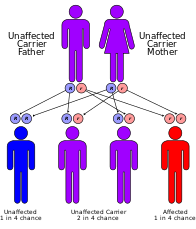
классифицируется Синдром Кокейна генетически следующим образом: [13]
| Тип | МОЙ БОГ | Ген |
|---|---|---|
| А | 216400 | ERCC8 (также называемый CSA) |
| Б | 133540 | ERCC6 (также называемый CSB) |
| С | 216411 | никто не известен |
- Мутации в гене ERCC8 (также известном как CSA) или гене ERCC6 (также известном как CSB) являются причиной синдрома Кокейна типа A и типа B. [8] Мутации гена ERCC6 составляют ~70% случаев. Белки, образуемые этими генами, участвуют в восстановлении поврежденной ДНК посредством механизма репарации, связанной с транскрипцией , особенно ДНК в активных генах. Повреждение ДНК вызвано ультрафиолетовыми лучами солнечного света, радиацией или свободными радикалами в организме. Нормальная клетка может восстановить повреждение ДНК до того, как оно накопится. Если ген ERCC6 или ERCC8 изменен (как при синдроме Кокейна), повреждения ДНК, возникшие во время транскрипции, не восстанавливаются, что приводит к остановке РНК-полимеразы в этом месте, что мешает экспрессии генов. По мере накопления невосстановленных повреждений ДНК все более активная экспрессия генов затрудняется, что приводит к нарушению работы клеток или их гибели, что, вероятно, способствует появлению таких признаков синдрома Кокейна, как преждевременное старение и гипомиелинизация нейронов. [8]
Механизм
[ редактировать ]В отличие от клеток с нормальной способностью к репарации, клетки с дефицитом CSA и CSB не способны преимущественно восстанавливать димеры циклобутан-пиримидина, индуцированные действием ультрафиолетового (УФ) света на матричную цепь активно транскрибируемых генов . [14] Этот дефицит отражает потерю способности выполнять процесс репарации ДНК, известный как эксцизионное восстановление нуклеотидов, связанное с транскрипцией (TC-NER). [15]
Внутри поврежденной клетки белок CSA обычно локализуется в местах повреждения ДНК , особенно в местах межцепочечных поперечных связей, двухцепочечных разрывах и некоторых моноаддуктах. [16] Белок CSB также обычно рекрутируется в участки поврежденной ДНК, и его рекрутирование происходит наиболее быстро и надежно следующим образом: межцепочечные сшивки > двухцепочечные разрывы > моноаддукты > окислительное повреждение. [16] Белок CSB образует комплекс с другим белком репарации ДНК, SNM1A ( DCLRE1A ), 5'-3'- экзонуклеазой , которая локализуется в межцепочечных поперечных связях транскрипционно-зависимым образом. [17] Накопление белка CSB в местах двухцепочечных разрывов ДНК происходит транскрипционно-зависимым образом и облегчает гомологичную рекомбинационную репарацию разрывов. [18] Во время фазы G0 / G1 клеточного цикла повреждение ДНК может запустить CSB-зависимый процесс рекомбинационной репарации, в котором используется матрица РНК (а не ДНК ). [19]
Особенности преждевременного старения при CS, вероятно, обусловлены, по крайней мере частично, нарушениями репарации ДНК (см. Теорию старения, связанную с повреждением ДНК ). [15]
Диагностика
[ редактировать ]У людей с этим синдромом размеры головы меньше нормальных ( микроцефалия ), низкий рост ( карликовость ), глаза кажутся запавшими, и они выглядят «стареющими». У них часто длинные конечности с контрактурами суставов (неспособность расслабить мышцы в суставах), сгорбленная спина ( кифоз ), а также они могут быть очень худыми ( кахетическими ) из-за потери подкожного жира. Их маленький подбородок, большие уши и заостренный тонкий нос часто придают им постаревший вид. [9] Кожа у людей с синдромом Коккейна также часто поражается: гиперпигментация, варикозное расширение вен или сосудистые звездочки ( телеангиэктазии ), [9] и серьезная чувствительность к солнечному свету распространены даже у людей без XP-CS. Часто у пациентов с синдромом Кокейна возникают сильные ожоги или образование волдырей при очень небольшом воздействии тепла.Глаза пациентов могут поражаться по-разному, и при КС часто встречаются глазные аномалии. Катаракта и помутнение роговицы ( помутнение роговицы ) являются обычным явлением. Может произойти потеря и повреждение нервов зрительного нерва, вызывающее атрофию зрительного нерва. [3] Нистагм , или непроизвольные движения глаз, и неспособность расширяющихся зрачков свидетельствуют о потере контроля над произвольными и непроизвольными движениями мышц. [9] Типичным признаком также является пигментация сетчатки в виде соли и перца.Диагноз устанавливается с помощью специального теста на репарацию ДНК, который измеряет восстановление РНК после воздействия УФ-излучения. Несмотря на то, что CS связан с генами, участвующими в эксцизионной репарации нуклеотидов (NER), в отличие от пигментной ксеродермы , CS не связан с повышенным риском развития рака. [6]
Лабораторные исследования
[ редактировать ]У пациентов с синдромом Кокейна в клетках, облученных УФ-излучением, наблюдается снижение синтеза ДНК и РНК. https://emedicine.medscape.com/article/1115866-workup#c5 Лабораторные исследования в основном полезны для устранения других нарушений. Например, рентгенография скелета, эндокринологические тесты и исследования хромосомных нарушений могут помочь исключить заболевания, включенные в дифференциальный диагноз. [ нужна ссылка ]
Исследования изображений
[ редактировать ]КТ головного мозга у пациентов с синдромом Коккейна может выявить кальцификацию и атрофию коры. [15]
Другие тесты
[ редактировать ]Возможна пренатальная оценка. Культивирование клеток амниотической жидкости используется для демонстрации того, что клетки плода испытывают дефицит синтеза РНК после УФ-облучения. [ нужна ссылка ]
Неврология
[ редактировать ]Визуализирующие исследования выявляют повсеместное отсутствие миелиновых оболочек нейронов белого вещества головного мозга и общую атрофию коры. [6] Кальцификации также были обнаружены в скорлупе — области переднего мозга , которая регулирует движения и помогает в некоторых формах обучения. [9] вместе с корой. [7] Кроме того, атрофия центральной области мозжечка , обнаруженная у пациентов с синдромом Коккейна, также может приводить к отсутствию мышечного контроля, особенно непроизвольному, и обычно наблюдаемой плохой осанке. [ нужна ссылка ]
Уход
[ редактировать ]Постоянного лечения этого синдрома не существует, хотя пациентов можно лечить симптоматически. Лечение обычно включает физиотерапию и небольшие операции на пораженных органах, например, удаление катаракты. [3] Также рекомендуется носить солнцезащитный крем с высоким фактором защиты и защитную одежду, поскольку пациенты с синдромом Коккейна очень чувствительны к ультрафиолетовому излучению. [20] Оптимальное питание также может помочь. Родителям рекомендуется генетическое консультирование, поскольку вероятность передачи заболевания будущим детям составляет 25%; также возможно пренатальное тестирование. [3] Другим важным аспектом является предотвращение рецидивов КС у других братьев и сестер. Выявление генетических дефектов позволяет предложить генетическое консультирование и дородовое наблюдение.диагностическое тестирование родителям, у которых уже есть один больной ребенок. [21]
В настоящее время реализуются два проекта, направленных на разработку генной терапии синдрома Кокейна. Первый проект, возглавляемый Ассоциацией детей с редкими заболеваниями Вильема Юлияна, направлен на разработку генной терапии специально для синдрома Коккейна типа B. [22] Второй проект, возглавляемый Riaan Research Initiative, посвящен разработке генной терапии синдрома Коккейна типа А. [23]
Прогноз
[ редактировать ]Прогноз для людей с синдромом Коккейна плохой , поскольку смерть обычно наступает в возрасте до 12 лет. [24] Прогноз синдрома Коккейна зависит от типа заболевания. Существует три типа синдрома Коккейна в зависимости от тяжести и начала симптомов. Однако различия между типами не всегда очевидны, и некоторые исследователи полагают, что признаки и симптомы отражают спектр, а не отдельные типы:Синдром Кокейна типа А (CSA) характеризуется нормальным развитием до тех пор, пока ребенку не исполнится 1 или 2 года, после чего рост замедляется и наблюдаются задержки развития. Симптомы не проявляются до 1 года. Ожидаемая продолжительность жизни при типе А составляет примерно от 10 до 20 лет. Эти симптомы наблюдаются у детей CS 1 типа.Синдром Кокейна типа B (CSB), также известный как «синдром церебро-окуло-фацио-скелетного (COFS)» (или «синдром Пена-Шокейра типа B»), является наиболее тяжелым подтипом. Симптомы присутствуют при рождении, а нормальное развитие мозга прекращается после рождения. Средняя продолжительность жизни детей с типом В — до 7 лет. Эти симптомы наблюдаются у детей 2-го типа CS.Синдром Кокейна типа C (CSC) появляется позже в детстве с более легкими симптомами, чем другие типы, и более медленным прогрессированием заболевания. Люди с этим типом синдрома Коккейна доживают до зрелого возраста, средняя продолжительность жизни составляет от 40 до 50 лет. Эти симптомы наблюдаются при CS типа 3. [15]
Эпидемиология
[ редактировать ]Синдром Кокейна редко встречается во всем мире. Расовая предрасположенность к синдрому Коккейна не выявлена. Для синдрома Коккейна не описано никаких сексуальных пристрастий; соотношение мужчин и женщин одинаково. Синдром Кокейна I (CS-A) манифестирует в детском возрасте. Синдром Кокейна II (CS-B) проявляется при рождении или в младенчестве и имеет худший прогноз. [15]
Недавние исследования
[ редактировать ]В недавнем исследовании, проведенном в январе 2018 года, упоминаются различные особенности CS, которые наблюдаются во всем мире и имеют сходства и различия:Заболеваемость КС составляет 1 на 250 000 живорождений, а распространенность — примерно 1 на 2,5 миллиона, что удивительно стабильно в различных регионах мира: [25] [26]
| Затронутые части | Клинические особенности | патология |
|---|---|---|
| Лицо | Высохшие лица. Запавшие глаза, большие уши, тонкий заостренный нос. Маленький подбородок. Кариес зубов , гипоплазия эмали. | |
| Кожа , волосы , ногти | Фоточувствительность . Кожа морщинистая и стареющая. Тонкие сухие волосы, преждевременная седина. Плохой венозный доступ. | |
| Центральная нервная система | Микроцефалия обычно начинается в возрасте 2 лет. Умственная отсталость с низким IQ . Отсроченные этапы . Тремор , атаксия , судороги , инсульты и субдуральные кровоизлияния . | Демиелинизация – очаговая и сегментарная – « Метахроматическая лейкодистрофия ». как олигодендроглии , так и шванновские клетки Поражаются . Поражается головного мозга белое вещество , мозолистое тело , ствол мозга , спинной мозг и периферические нервы . Гибель нейронов во многих местах, особенно в мозжечке . Потеря клеток переднего рога вследствие антероградной и/или ретроградной дегенерации . Кальцификация [55–95%] коры головного мозга (особенно глубины борозд , базальных ганглиев , мозжечка, таламуса , а также артерий , артериол и капилляров ) . Сосудистые изменения - Струнные сосуды , особенно в участках метахроматической лейкодистрофии, кальциноз в лептоменингеальных сосудах, ускоренный атеросклероз и атеролосклероз . Глиоз присутствует. Астроциты и микроглия могут иметь неправильную цитоплазму , множественные ядра . может наблюдаться как белое вещество высокой интенсивности FLAIR MRI На сигналах последовательностей . головного мозга нет Серьезных пороков развития . Отмечается относительная сохранность коры головного мозга, небольшое истончение корковой ленты. Нормальный рисунок извилин с расширением борозд . Слоистость, размер нейронов и конфигурация неокортекса сохранены . Может проявляться теменно- затылочное доминирование. мозжечка Тяжелая атрофия . Потеря Пуркинье , зернистых нейронов и в некоторых случаях нейронов зубчатого ядра . Дендриты клеток Пуркинье могут быть сильно деформированными («цветки кактуса»), ожелезненными дендритами. Дендриты имеют меньше ветвей более высокого порядка. « аксональные торпеды Могут присутствовать » Пуркинье. увеличение желудочков , увеличенная большая цистерна Отмечается . Амилоидные бляшки , нейрофибриллярные клубки , тельца Хирано обычно не наблюдаются, хотя убиквитиновая реактивность аксонов . присутствует |
| Слух и вестибулярная система | Нейросенсорная на высокие тона потеря слуха [60–90 %]. Смешанная кондуктивная и нейросенсорная тугоухость (44%) Чаще всего двусторонняя, реже односторонняя. | Потеря волосковых клеток в улитке , особенно в базальном обороте . Гибель нейронов спирального ганглия . Атрофия слуховых путей . Scala communis , стремечко утолщено расширен , прототимпанум . Потеря волосковых клеток в верхней части. Гибель нейронов вестибулярного ганглия . Коллапс эндолимфатического протока нижней части тела |
| Зрение | Помутнение роговицы . Катаракта [36–86%]. Обычно двустороннее, большинство развивается к 4 годам. Пигментная ретинопатия («соль и перец») [43–89%]. Миотические зрачки , диска зрительного нерва бледность , энофтальм , узкие глазные щели . | Очаговая потеря меланина пигмента гранул . Отложение липофусцина , крупные пигментированные клетки в периваскулярном распределении. сетчатки пигментного эпителия Атрофия и гиперплазия . Потеря клеток в ганглиях и наружных слоях ядерных клеток. как внешние, так и внутренние сегменты фоторецепторов Поражаются зрительного нерва . Атрофия с частичной демиелинизацией , потерей аксонов и глиозом. |
| Опорно-двигательная система | Кахектическая карликовость . Контрактуры . Кифоз , сколиоз . Сгорбленная поза. Мышечная атрофия . | Денервационная миопатия , атрофия без использования |
| Сердечно-сосудистая система | Ускоренная гипертензия . Расширение корня аорты . Кардиомиопатия . | Повышенное утолщение интимы- медии. Атеросклероз , артериосклероз . |
| Желудочно-кишечная система | Тяжелый рефлюкс . Нарушение перистальтики желудочно-кишечного тракта . У многих есть чрескожные гастростомические трубки . Гепатомегалия , спленомегалия , повышение активности печеночных ферментов . Измененный метаболизм лекарств. | - |
| Почечная система | Почечная недостаточность | В почечных артериях наблюдаются изменения при прогрессирующем атеросклерозе и артериолосклерозе. Односторонние или гипоплазированные почки . |
| Репродуктивная система | - | - |
| Мужчины | Микропенис , меньший яичек размер | - |
| Женщины | Атрофия яичников . успешной беременности . Сообщается об | - |
| Эндокринная система | Нормальные вторичные половые признаки . Нормальный гормона роста , тиреотропного гормона , кальция. уровень | Нормальный гипофиз и щитовидная железа |
| Эккринные системы | Снижение выработки пота , слез , слюны. | - |
См. также
[ редактировать ]- Болезнь ускоренного старения
- Биогеронтология
- Дегенеративное заболевание
- Генетическое заболевание
- Синдром CAMFAK — считается формой (или подмножеством) синдрома Коккейна. [27]
Ссылки
[ редактировать ]- ^ Бертола; Цао, Х; Альбано, Lm; Оливейра, доктор медицинских наук; Кок, Ф; Маркес-Диас, Mj; Ким, Калифорния; Гегеле, Ра (2006). «Синдром Коккейна типа А: новые мутации у восьми типичных пациентов» . Журнал генетики человека . 51 (8): 701–5. дои : 10.1007/s10038-006-0011-7 . ПМИД 16865293 .
- ^ Джеймс, Уильям; Бергер, Тимоти; Элстон, Дирк (2005). Кожные заболевания Эндрюса: клиническая дерматология (10-е изд.). Сондерс. п. 575 . ISBN 978-0-7216-2921-6 .
- ^ Перейти обратно: а б с д и Бендер М., Потоцкий Л., Метри Д. Что это за синдром? Синдром Кокейна. Детская дерматология [сериал онлайн]. Ноябрь 2003 г.;20(6):538-540. Доступно по адресу: MEDLINE с полным текстом, Ипсвич, Массачусетс. По состоянию на 30 апреля 2015 г.
- ^ Лаугель, Винсент (2013). «Синдром Коккейна: расширяющийся клинический и мутационный спектр» . Механизмы старения и развития . 134 (5–6): 161–170. дои : 10.1016/j.mad.2013.02.006 . ISSN 1872-6216 . ПМИД 23428416 . S2CID 19137836 .
- ^ Хоймейкерс Дж. Х. (октябрь 2009 г.). «Повреждение ДНК, старение и рак». Н. англ. Дж. Мед . 361 (15): 1475–85. дои : 10.1056/NEJMra0804615 . ПМИД 19812404 .
- ^ Перейти обратно: а б с д Нэнс М., Берри С. (1 января 1992 г.). «Синдром Коккейна: обзор 140 случаев». Американский журнал медицинской генетики . 42 (1): 68–84. дои : 10.1002/ajmg.1320420115 . ПМИД 1308368 .
- ^ Перейти обратно: а б с д Нил Калифорния, Дингуолл ММ. Синдром, напоминающий прогерию: обзор двух случаев. Архив болезней в детстве. 1950;25(123):213-223.
- ^ Перейти обратно: а б с д и ж Синдром Кокейна. Домашний справочник по генетике http://ghr.nlm.nih.gov/condition/cockayne-syndrome Опубликовано 28 апреля 2015 г. Проверено в мае 2010 г. По состоянию на 30 апреля 2015 г.
- ^ Перейти обратно: а б с д и ж Джавадзаде М. Синдром Кокейна. Иран Дж. Чайлд Нейрол. Осень 2014 г.;8;4(Приложение 1):18-19.
- ^ Цереброокуло-фациоскелетный синдром 2. Менделевское наследование онлайн у человека. https://omim.org/entry/610756 . Опубликовано 12.02.2007.
- ^ Синдром Лаугеля В. Кокейна. 28 декабря 2000 г. [Обновлено 14 июня 2012 г.]. В: Пагон Р.А., Адам М.П., Ардингер Х.Х. и др., редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2015 гг. Доступно: [1]
- ^ «Повреждение клетки и смерть» . Библиотека медицинских концепций Lecturio . Проверено 7 июля 2021 г.
- ^ Вессони, Александр Тейшейра; Герра, Камила Чавес Коэльо; Кадзитани, Густаво Сатору; Насименто, Ливия Лус Соуза; Гарсия, Камила Карриан Мачадо (2020). «Синдром Коккейна: множество проблем и подходов к пониманию многогранного заболевания» . Генетика и молекулярная биология . 43 (1 доп. 1): e20190085. дои : 10.1590/1678-4685-GMB-2019-0085 . ISSN 1415-4757 . ПМК 7250278 . ПМИД 32453336 .
- ^ ван Хоффен А., Натараджан А.Т., Мейн Л.В., ван Зеланд А.А., Маллендерс Л.Х., Венема Дж. (1993). «Недостаточная репарация транскрибируемой цепи активных генов в клетках синдрома Кокейна» . Нуклеиновые кислоты Рез . 21 (25): 5890–5. дои : 10.1093/нар/21.25.5890 . ПМК 310470 . ПМИД 8290349 .
- ^ Перейти обратно: а б с д и Кариккинет А., Шайби-Кнудсен М., Файвенсон Э., Крото Д., Бор Б. (январь 2017 г.). «Синдром Коккейна: клинические особенности, модельные системы и пути» . Обзоры исследований старения . 33 : 3–17. дои : 10.1016/J.arr.2016.08.002 . ПМК 5195851 . ПМИД 27507608 .
- ^ Перейти обратно: а б Ияма Т., Уилсон Д.М. (2016). «Элементы, которые регулируют реакцию белков, дефектных при синдроме Кокейна, на повреждение ДНК» . Дж. Мол. Биол . 428 (1): 62–78. дои : 10.1016/j.jmb.2015.11.020 . ПМЦ 4738086 . ПМИД 26616585 .
- ^ Ияма Т., Ли С.Ю., Берквист Б.Р., Гилеади О., Бор В.А., Зейдман М.М., МакХью П.Дж., Уилсон Д.М. (2015). «CSB взаимодействует с SNM1A и способствует процессингу межцепочечных сшивок ДНК» . Нуклеиновые кислоты Рез . 43 (1): 247–58. дои : 10.1093/nar/gku1279 . ПМЦ 4288174 . ПМИД 25505141 .
- ^ Батенбург Н.Л., Томпсон Э.Л., Хендриксон Э.А., Чжу XD (2015). «Белок группы B синдрома Коккейна регулирует восстановление двухцепочечных разрывов ДНК и активацию контрольных точек» . ЭМБО Дж . 34 (10): 1399–416. дои : 10.15252/embj.201490041 . ПМК 4491999 . ПМИД 25820262 .
- ^ Вэй Л., Накадзима С., Бём С., Бернштейн К.А., Шен З., Цанг М., Левин А.С., Лан Л. (2015). «Повреждение ДНК во время фазы G0/G1 запускает РНК-матрицу, синдром Коккейна B-зависимую гомологичную рекомбинацию» . Учеб. Натл. акад. наук. США . 112 (27): E3495–504. Бибкод : 2015PNAS..112E3495W . дои : 10.1073/pnas.1507105112 . ПМК 4500203 . ПМИД 26100862 .
- ^ Киллермен, Мартен. Синдром Кокейна. Шведский информационный центр редких заболеваний. 2012: 4.0. http://www.socialstyrelsen.se/rarediseases/cockaynesyndrome#anchor_17 Архивировано 24 сентября 2015 г. в Wayback Machine.
- ^ Название: Синдром Кокейна Авторы: доктор Нита Р. Сутай, доктор медицины Ашфак Тинмасвала, доктор Манджири Карлекар, доктор Свати Джа http://jmscr.igmpublication.org/v3-i7/35%20jmscr.pdf
- ^ админ (23.10.2020). «Синдром Коккейна типа b | Ассоциация Вильема Юлияна» . Лечение синдрома Кокейна – тип B. Проверено 6 июля 2023 г.
- ^ «Исследовательская инициатива Риаана — синдром Кокейна» . Проверено 6 июля 2023 г.
- ^ «Синдром Коккейна | Информационный центр по генетическим и редким заболеваниям (GARD) – программа NCATS» .
- ^ Кариккинет, AC; Шайби-Кнудсен, М.; Файвенсон, Э.; Крото, ДЛ; Бор, Вирджиния (2016). «Синдром Коккейна: клинические особенности, модельные системы и пути» . Обзоры исследований старения . 33 : 3–17. дои : 10.1016/J.arr.2016.08.002 . ПМК 5195851 . ПМИД 27507608 .
- ^ Кубота, Масая; Охта, Саяка; Андо, Аки; Кояма, Акико; Терашима, Хироши; Касии, Хирофуми; Хосино, Хидеки; Сугита, Кацуо; Хаяси, Масахару (июнь 2015 г.). «Общенациональное исследование синдрома Кокейна в Японии: заболеваемость, клиническое течение и прогноз: синдром Коккейна в Японии» . Международная педиатрия . 57 (3): 339–347. дои : 10.1111/пед.12635 . ПМИД 25851792 . S2CID 5311897 .
- ^ «Орфанет: синдром CAMFAK» .
Внешние ссылки
[ редактировать ]- Эта статья включает в себя некоторые общедоступные тексты из Национальной медицинской библиотеки США.