Акроцефалосиндактилия
| Акроцефалосиндактилия | |
|---|---|
| Другие имена | СКУД |
 | |
| Синдактилия при акроцефалосиндактилии (Аперта) | |
| Специальность | Медицинская генетика |
Акроцефалосиндактилия — группа врожденных заболеваний, характеризующихся неправильными чертами лица и черепа ( краниосиностоз ), а также кистей и стоп ( синдактилия ). [ 1 ] Краниосиностоз возникает, когда черепные швы, волокнистая ткань, соединяющая кости черепа, срастаются с черепными костями на ранних стадиях развития. Черепные швы позволяют костям черепа продолжать расти до тех пор, пока они не срастутся в возрасте 24 лет. Преждевременное сращение черепных швов может привести к изменению формы черепа и препятствовать росту мозга. [ 2 ] [ 3 ] Синдактилия возникает, когда пальцы рук или ног слиты вместе. [ 4 ] Если также присутствует полидактилия , классифицируется как акроцефалополисиндактилия. [ 5 ] Полидактилия возникает, когда на руках или ногах имеются дополнительные пальцы. [ 6 ] Акроцефалосиндактилия обычно диагностируется после рождения, хотя иногда возможна пренатальная диагностика, если генетическая вариация присутствует у членов семьи, поскольку заболевания обычно наследуются по аутосомно-доминантному типу. [ 1 ] Лечение часто включает хирургическое вмешательство в раннем детстве для коррекции краниосиностоза. [ 7 ] и синдактилия . [ 8 ]
Тяжесть симптомов акроцефалосиндактилии значительно варьируется в зависимости от подтипа и лечения на ранних этапах жизни.
История
[ редактировать ]Случаи этого заболевания были зарегистрированы еще в 18 веке. [ 9 ] Термин акроцефалосиндактилия (от греческого ἄκρος ( ákros) «самый высокий, на конечности», κεφαλή (kephaloḗ) «голова», σύν (син) «вместе» и δάκτυλος (дактилос) «палец») был впервые применен в 1906 году французским врачом. Эжен Апер первым описал состояние, характеризующееся краниосиностозом и синдактилией . [ 10 ] Состояние, описанное Апертом, теперь известно как подтип синдрома Аперта — акроцефалосиндактилия. [ 11 ] На протяжении ХХ века характеризовались и другие подтипы акроцефалосиндактилии. [ 1 ]
Распространенность
[ редактировать ]Учитывая все виды акроцефалосиндактилии, на каждые 65 000 - 102 500 рожденных детей рождается один новорожденный ребенок с акроцефалосиндактилией. Нет разницы в количестве мужчин и женщин, страдающих акроцефалосиндактилией. [ 12 ] [ 13 ] [ 14 ] [ 15 ]
Характеристики
[ редактировать ]Акроцефалосиндактилия проявляется во многих различных подтипах, однако наблюдается значительное совпадение симптомов. Как правило, все формы акроцефалосиндактилии характеризуются атипичными чертами черепа, кистей и стоп, такими как преждевременное закрытие фиброзных соединений между определенными костями черепа, [ 16 ] [ 17 ] слияние определенных пальцев рук или ног, [ 16 ] [ 18 ] и/или больше обычного количества цифр. [ 19 ] Некоторые подтипы также включают структурные изменения сердца , присутствующие при рождении. [ 20 ] [ 1 ]
Причина
[ редактировать ]Большинство форм акроцефалосиндактилии или акроцефалополисиндактилии наследуются по аутосомно-доминантному типу. [ 15 ] [ 20 ] [ 1 ] за исключением синдрома Карпентера , который наследуется по аутосомно-рецессивному типу. de-novo Сообщалось, что варианты или генетические изменения, не унаследованные от родителей, в различных генах вызывают несколько типов акроцефалосиндактилии. Среди известных генетических изменений есть вариации в таких генах, как рецептор фактора роста фибробластов ( FGFR ) , [ 16 ] [ 17 ] [ 21 ] [ 22 ] ТВИСТ1 , [ 23 ] и РАБ23 . [ 24 ] Конститутивная активация этих категорий генов, особенно FGF R, который активно участвует на стадиях развития эмбрионов, таких как развитие органов или органогенез , а также поддержание тканеобразующих клеток, известных как клетки-предшественники , может быть очень вредной. [ 25 ]
Все генетически унаследованные состояния акроцефалосиндактилии демонстрируют пенетрантность от высокой до полной с различной выраженностью , что означает, что у всех людей, унаследовавших это состояние, наблюдаются атипичные характерные структуры черепа, рук и ног, но тяжесть нарушений варьируется. [ 1 ] В некоторых случаях повышенный возраст отца считается фактором риска. [ 1 ]
Влияние условий на жизнь
[ редактировать ]Несмотря на текущие значительные усилия хирургического лечения последствий акроцефалосиндактилии, заболеваемость все еще существует у людей, получивших лечение. Те, кто достигает совершеннолетия, часто имеют более низкий уровень образования, чем их сверстники, а также большие трудности в различных социальных аспектах, таких как свидания, брак или сексуальные отношения. сообщать о необходимости проживания с уходом на протяжении всей жизни, а также о других проблемах со здоровьем, таких как проблемы со слухом или эпилепсия . Они также могут чаще, чем их сверстники, [ 26 ]
К счастью, многие люди с этим заболеванием сообщают о таком же уровне счастья в своей жизни, как и люди, не страдающие этим заболеванием. [ 26 ] и продемонстрировать высокую социальную интеграцию , а также большую физическую и эмоциональную устойчивость, несмотря на любые препятствия. [ 27 ]
Диагностика
[ редактировать ]Пренатальная диагностика
[ редактировать ]до рождения Диагностика некоторых форм акроцефалосиндактилии возможна . Пренатальная генетическая диагностика возможна только в том случае, если известна вариация гена, ответственная за синдром, и вариация, вызывающая заболевание, идентифицирована в геноме члена семьи. Сбор образцов для генетического тестирования можно провести с помощью амниоцентеза , при котором берутся эмбриональные стволовые клетки, содержащиеся в околоплодных водах, или отбора проб ворсинок хориона , при котором берутся образцы плацентарных клеток. [ 15 ] Известен случай пренатальной диагностики синдрома Аперта с помощью фетоскопии , при которой плод наблюдают с помощью эндоскопа, введенного в матку из брюшной полости. [ 22 ] Альтернативно, существует интерес к использованию неинвазивных методов, таких как ультразвук, для обнаружения атипичных особенностей черепа плода. [ 28 ]
Послеродовая диагностика
[ редактировать ]Большинство диагнозов акроцефалосиндактилии ставятся после рождения путем оценки физических симптомов младенца. Это можно подтвердить с помощью рентгенографических изображений, таких как рентгеновские снимки, и молекулярно -генетического тестирования , которое ищет вариации ДНК, которые, как известно, вызывают заболевание. [ 29 ] [ 30 ] Молекулярно-генетическое тестирование обычно проводится в генах FGFR , TWIST1 и RAB23 .
Номенклатура/Классификация
[ редактировать ]Не существует единой номенклатуры или классификации различных синдромов, относящихся к акроцефалосиндактилии и акроцефалополисиндактилии. Хотя об акроцефалосиндактилии сообщалось еще в XVIII веке, [ 9 ] Классификации ACS и ACPS появились только во второй половине 20 века. Однако эта классификация может быть устаревшей, поскольку было предложено стереть различие между акроцефалосиндактилией и акроцефалополисиндактилией. [ 5 ]
В настоящее время синдром Ноака (ACPS типа I) теперь классифицируется как синдром Пфайффера (ACS типа V); [ 31 ] Синдром Гудмана (тип ACPS IV) классифицируется как разновидность синдрома Карпентера (тип ACPS II); [ 19 ] и разные исследователи объединили синдром Аперта (ASC типа I), Крузона (ASC типа II) и Пфайффера (ASC типа V) в синдром Аперта-Крузона. [ 32 ] и Крузон-Пфайффер [ 33 ] синдром.
Акроцефалосиндактилия IV типа раньше называлась синдромом Мора, однако позже была отнесена к орофацио-дигитальному синдрому типа II. [ 34 ] Синдром Пфайффера ранее относился к типу VI и типу Ваарденбурга V, но это изменилось где-то после 1966 года. [ 35 ]
Акроцефалосиндактилия (АКС):
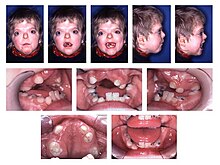
- тип I – синдром Аперта [ 11 ]
- тип II – синдром Крузона [ 36 ]
- тип III - синдром Сэтре-Чотцена [ 37 ]
- Синдром Робинова-Сорауфа предложено включить в классификацию Сэтре-Чотцена. [ 38 ]
- тип IV – синдром Мора (архаичный) [ 34 ] [ 35 ]
- тип V – синдром Пфайффера [ 31 ]
- Синдром Ноака включен в классификацию синдрома Пфайффера [ 31 ]
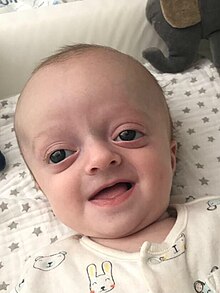
Родственный термин, акроцефалополисиндактилия (ACPS), относится к включению полидактилии в предлежание. Он также имеет несколько типов:
- тип I – синдром Ноака (архаичный) [ 31 ]
- тип II – синдром Карпентера [ 39 ]
- Синдром Гудмана включен в синдрома Карпентера классификацию [ 40 ]
- Синдром Саммит включен в синдрома Карпентера классификацию [ 41 ]
- тип III – синдром Сакати-Нихана-Тисдейла [ 42 ]
- тип IV – синдром Гудмана (архаичный) [ 40 ]
Уход
[ редактировать ]Краниосиностоз
[ редактировать ]При подтипах с краниосиностозом требуется хирургическое вмешательство для предотвращения преждевременного сращения черепных швов, например венечного шва ( брахицефалия ). [ 7 ] Черепной шов, расположенный между двумя лобными и двумя теменными костями черепа, называется венечным швом. [ 2 ] Краниопластику следует выполнять на первом году жизни для предотвращения нарушений роста мозга из-за повышения внутричерепного давления. Операция на средней зоне лица также может потребоваться в детстве, чтобы отделить среднюю часть лица от остальной части черепа для устранения респираторных и ортодонтических проблем. [ 43 ] [ 44 ]
Синдактилия
[ редактировать ]Синдактилия при некоторых подтипах редко бывает настолько серьезной, чтобы повлиять на функцию рук, поэтому лечение может не потребоваться. [ 43 ]
При более тяжелых подтипах, таких как синдром Аперта , может потребоваться хирургическая коррекция синдактилии. Операцию рекомендуется проводить как можно раньше, обычно в возрасте 4 месяцев. Лечение зависит от тяжести синдактилии. Хирургическое лечение обычно включает освобождение межпальцевой перепонки и удлинение большого пальца. [ 8 ]
Управление
[ редактировать ]Лечение людей с диагнозом акроцефалосиндактилия выходит за рамки хирургического вмешательства. Есть много шагов, которые могут помочь в долгосрочном лечении синдрома. Лица, страдающие акроцефалосиндактилией, и лица, осуществляющие уход за ними, могут построить систему поддержки здравоохранения, построив прочные отношения с командой медицинских специалистов. Предварительно сформированные группы медицинских специалистов часто можно встретить в университетах или научно-исследовательских институтах . Лица, осуществляющие уход, могут предотвратить будущие проблемы, изучая варианты финансовой помощи , медицинского страхования и размещения в образовательных учреждениях. Лицам, осуществляющим первичный уход, рекомендуется уделять приоритетное внимание своему эмоциональному здоровью, выделяя время для себя и находя надежную систему поддержки. [ 29 ]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Перейти обратно: а б с д и ж г Биссоннетт, Бруно; Лугинбюль, Игорь; Марчиняк, Бруно; Даленс, Бернард Дж. (2006), «Синдромы акроцефалосиндактилии» , Синдромы: быстрое распознавание и периоперационные последствия , Нью-Йорк, Нью-Йорк: Компании McGraw-Hill.
- ^ Перейти обратно: а б Рассел, Уильям П.; Рассел, Марк Р. (2023), «Анатомия, голова и шея, корональный шов» , StatPearls , Остров сокровищ (Флорида): StatPearls Publishing, PMID 30252267 , получено 4 декабря 2023 г.
- ^ Опперман, Луизиана (2000). «Черепные швы как места внутримембранозного роста костей» . Динамика развития . 219 (4): 472–485. doi : 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F . ISSN 1058-8388 . ПМИД 11084647 . S2CID 8801611 .
- ^ Малик, С. (2012). «Синдактилия: фенотипы, генетика и современная классификация» . Европейский журнал генетики человека . 20 (8): 817–824. дои : 10.1038/ejhg.2012.14 . ISSN 1476-5438 . ПМК 3400728 . ПМИД 22333904 .
- ^ Перейти обратно: а б Коэн, М. Майкл; Крейборг, Свен (май 1995 г.). «Руки и ноги при синдроме Аперта» . Американский журнал медицинской генетики . 57 (1): 82–96. дои : 10.1002/ajmg.1320570119 . ISSN 0148-7299 . ПМИД 7645606 .
- ^ Малик, С. (2014). «Полидактилия: фенотипы, генетика и классификация» . Клиническая генетика . 85 (3): 203–212. дои : 10.1111/cge.12276 . ISSN 0009-9163 . ПМИД 24020795 . S2CID 22412404 .
- ^ Перейти обратно: а б Краниосиностоз: диагностика, оценка и лечение . М. Майкл Коэн, Рут Э. Маклин (2-е изд.). Нью-Йорк: Издательство Оксфордского университета. 2000. ISBN 0-19-511843-Х . OCLC 41528658 .
{{cite book}}: CS1 maint: другие ( ссылка ) - ^ Перейти обратно: а б Рапосо-Амарал, Кассио Эдуардо; ДЕНАДАИ, Рафаэль; ФУРЛАН, Педро; Рапосо-Амарал, Сезар Аугусто (октябрь 2018 г.). «Лечение синдрома крепкой руки: стратегии достижения пятирукости» . Пластическая и реконструктивная хирургия . 142 (4): 972–982. дои : 10.1097/PRS.0000000000004815 . ISSN 0032-1052 . ПМИД 29994846 . S2CID 51614940 .
- ^ Перейти обратно: а б Уитон, Юго-Запад (1894 г.). «Два образца врожденной деформации черепа у младенцев, связанной со сращением пальцев рук и ног». Транс Патол Сок Лондон . 45 : 238–241.
- ^ Аперт, МЭ (1906). «Акроцефалосиндактилия». Бык. Память Соц. Мед. Прыгать. Париж . 23 : 13:10–13:30.
- ^ Перейти обратно: а б «Запись — #101200 — Синдром Аперта» . omim.org . Проверено 4 декабря 2023 г.
- ^ М. Дас, Джо; Уинтерс, Райан (2023), «Синдром Пфайффера» , StatPearls , Остров сокровищ (Флорида): StatPearls Publishing, PMID 30422477 , получено 4 декабря 2023 г.
- ^ Конради, Кристофер Д.; Патель, Бхупендра К.; Шарма, Сандип (2023 г.), «Синдром Аперта» , StatPearls , Остров сокровищ (Флорида): StatPearls Publishing, PMID 30085535 , получено 4 декабря 2023 г.
- ^ Толарова, М.М. (2023). «Генетика синдрома Крузона» . Педиатрия: генетика и метаболические заболевания – через Medscape.
- ^ Перейти обратно: а б с Галлахер, Эмили Р.; Ратисунторн, Чутима; Каннингем, Майкл Л. (1993), Адам, Маргарет П.; Фельдман, Джерри; Мирзаа, Гайда М.; Пагон, Роберта А. (ред.), «Синдром Сэтре-Чотцена» , GeneReviews® , Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл, PMID 20301368 , получено 4 декабря 2023 г.
- ^ Перейти обратно: а б с Уилки, Эндрю О.М.; Слейни, Сара Ф.; Олдридж, Майкл; Пул, Майкл Д.; Эшворт, Джеральдин Дж.; Хокли, Энтони Д.; Хейворд, Ричард Д.; Дэвид, Дэвид Дж.; Пуллейн, Луиза Дж.; Ратленд, Пол; Малькольм, Сьюзен; Винтер, Робин М.; Рирдон, Уильям (февраль 1995 г.). «Синдром Аперта возникает в результате локализованных мутаций FGFR2 и является аллелем синдрома Крузона» . Природная генетика . 9 (2): 165–172. дои : 10.1038/ng0295-165 . ISSN 1546-1718 . ПМИД 7719344 . S2CID 12423131 .
- ^ Перейти обратно: а б Каринчи, Франческо; Пеццетти, Фурио; Лоччи, Паола; Беккетти, Эннио; Карлс, Фридрик; Преимущество, Анна; Беккетти, Алессио; Каринчи, Паоло; Барони, Тициан; Бодо, Мария (май 2005 г.). «Синдромы Аперта и Крузона: клинические данные, гены и внеклеточный матрикс» . Журнал черепно-лицевой хирургии . 16 (3): 361–368. дои : 10.1097/01.SCS.0000157078.53871.11 . ISSN 1049-2275 . ПМИД 15915098 . S2CID 23327865 .
- ^ Биссоннетт, Бруно; Лугинбюль, Игорь; Энгельхардт, Томас (2019), «Акроцефалополисиндактилия типа IV: синдром Гудмана» , Синдромы: быстрое распознавание и периоперационные последствия (2-е изд.), Нью-Йорк, Нью-Йорк: McGraw-Hill Education
- ^ Перейти обратно: а б Коэн, Дональд М.; Грин, Джеймс Г.; Миллер, Дженис; Горлин, Роберт Дж.; Рид, Джерри А.; Опитц, Джон М.; Рейнольдс, Джеймс Ф. (октябрь 1987 г.). «Акроцефалополисиндактилия типа II — синдром Карпентера: клинический спектр и попытка объединения с синдромами Гудмана и Саммита» . Американский журнал медицинской генетики . 28 (2): 311–324. дои : 10.1002/ajmg.1320280208 . ISSN 0148-7299 . ПМИД 3322002 .
- ^ Перейти обратно: а б Кумар, Нирадж; Арора, Шубханги; Биндра, Ашиш; Гоял, Кешав (2014). «Анестетическое обеспечение лечения краниосиностоза у пациента с синдромом Аперта» . Саудовский журнал анестезии . 8 (3): 399–401. дои : 10.4103/1658-354X.136631 . ISSN 1658-354X . ПМЦ 4141395 . ПМИД 25191197 .
- ^ Гэлвин, Б.Д.; Харт, КК; Мейер, АН; Вебстер, МК; Донохью, диджей (23 июля 1996 г.). «Конститутивная активация рецептора мутациями синдрома Крузона в рецепторе фактора роста фибробластов (FGFR)2 и химерах FGFR2/Neu» . Труды Национальной академии наук . 93 (15): 7894–7899. Бибкод : 1996PNAS...93.7894G . дои : 10.1073/pnas.93.15.7894 . ISSN 0027-8424 . ПМЦ 38845 . ПМИД 8755573 .
- ^ Перейти обратно: а б Леонард, Клэр О.; Дайкоку, Норман Х.; Винн, Кевин; Опитц, Джон М. (январь 1982 г.). «Пренатальная фетоскопическая диагностика синдрома Аперта» . Американский журнал медицинской генетики . 11 (1): 5–9. дои : 10.1002/ajmg.1320110103 . ISSN 0148-7299 . ПМИД 7065003 .
- ^ Цай, Цзюаньлян; Гудман, Барбара К.; Патель, Анкита С.; Малликен, Джон Б.; Ван Малдергем, Лайонел; Хогансон, Джордж Э.; Пазнекас, Уильям А.; Бен-Нерия, Зива; Шеффер, Рут; Каннингем, Майкл Л.; Даентл, Донна Л.; Джабс, Этилин Ван (1 декабря 2003 г.). «Повышенный риск задержки развития при синдроме Сэтре-Чотцена связан с делециями TWIST: улучшенная стратегия скрининга мутаций TWIST» . Генетика человека . 114 (1): 68–76. дои : 10.1007/s00439-003-1012-7 . ISSN 1432-1203 . ПМИД 14513358 . S2CID 20929600 .
- ^ Дженкинс, Даган; Зеелов, Доминик; Джехи, Фернанда С.; Перлин, Чад А.; Алонсо, Луис Г.; Буэно, Даниэла Ф.; Доннаи, Дайан; Иосифьева, Драгана; Матийссен, Ирен М.Дж.; Мортон, Дженни EV; Орставик, Карен Хелен; Суини, Элизабет; Уолл, Стивен А.; Марш, Джеффри Л.; Нюрнберг, Питер (1 июня 2007 г.). «Мутации RAB23 при синдроме Карпентера подразумевают неожиданную роль передачи сигналов Hedgehog в развитии черепных швов и ожирении» . Американский журнал генетики человека . 80 (6): 1162–1170. дои : 10.1086/518047 . ISSN 0002-9297 . ПМК 1867103 . ПМИД 17503333 .
- ^ Ямадзи, Дзюмпей; Ватанабэ, Цукаса; Накатоми, Мицусиро; Оно, Кентаро, Кейджи; Кавамото, Тацуо (ноябрь 2018 г.) . синдрома аперта» . Динамика развития . 247 (11): 1175–1185. : 10.1002 /dvdy.24673 . ISSN 1058-8388 . PMID 30251381. doi S2CID 52815441 .
- ^ Перейти обратно: а б Товетьярн, Роберт; Тарнов, Питер; Мальтийский, Джованни; Фишер, Сара; Сахлин, Пер-Эрик; Кёлби, Ларс (октябрь 2012 г.). «Дети с синдромом Аперта во взрослом возрасте: последующее исследование 28 пациентов из Скандинавии» . Пластическая и реконструктивная хирургия . 130 (4): 572e–576e. дои : 10.1097/PRS.0b013e318262f355 . ISSN 0032-1052 . ПМИД 23018718 . S2CID 45847015 .
- ^ Тагиния, Амир Х.; Йорлетс, Рэйчел Р.; Дойл, Майкл; Лабоу, Брайан И.; Аптон, Джозеф (апрель 2019 г.). «Отдаленные функциональные результаты верхних конечностей у взрослых с синдромом Аперта» . Пластическая и реконструктивная хирургия . 143 (4): 1136–1145. дои : 10.1097/PRS.0000000000005479 . ISSN 0032-1052 . ПМИД 30676503 . S2CID 59225959 .
- ^ «Энциклопедия генетических нарушений и врожденных дефектов | WorldCat.org» . search.worldcat.org . Проверено 4 декабря 2023 г.
- ^ Перейти обратно: а б «Синдром Аперта - Диагностика и лечение - Информационный центр генетических и редких заболеваний» . Rarediseases.info.nih.gov . Проверено 5 ноября 2022 г.
- ^ Гонсалес, М.; Хойерц, С.; Мартинович, Дж.; Делахай, С.; Базен, А.; Логет, П.; Паскье, Л.; Ле Меррер, М.; Бонавентура, Дж. (17 июня 2005 г.). «Аномалии позвоночника и хрящевой рукав трахеи у трех пациентов с синдромом Пфайффера, несущих мутацию S351C FGFR2: письмо в редакцию» . Клиническая генетика . 68 (2): 179–181. дои : 10.1111/j.1399-0004.2005.00477.x . ПМИД 15996217 . S2CID 1652216 .
- ^ Перейти обратно: а б с д «Запись № 101600 — Синдром Пфайффера» . omim.org . Проверено 4 декабря 2023 г.
- ^ Редей, Джордж П., изд. (2008), «Синдром Аперта или Аперта-Крузона» , Энциклопедия генетики, геномики, протеомики и информатики , Дордрехт: Springer Нидерланды, стр. 127, номер домена : 10.1007/978-1-4020-6754-9_1009 , ISBN 978-1-4020-6754-9 , получено 5 ноября 2022 г.
- ^ Уилсон, Александр Т.; де Планк, Катрин А.; Ян, Сумин С.; Таскер, Роберт С.; ван Веелен, Мари-Лиза К.; Дреммен, Маржолейн Х.Г.; Вруман, Анри А.; Матийссен, Ирен MJ (октябрь 2020 г.). «Толщина коры при синдроме Крузона-Пфайффера: данные, касающиеся первичного расширения свода черепа» . Пластическая и реконструктивная хирургия. Глобальный открытый турнир . 8 (10): е3204. дои : 10.1097/GOX.0000000000003204 . ISSN 2169-7574 . ПМЦ 7647527 . ПМИД 33173703 .
- ^ Перейти обратно: а б «Запись — %252100 — Синдром Мора» . omim.org . Проверено 5 ноября 2022 г.
- ^ Перейти обратно: а б МакКьюсик, Вирджиния (1966). «Аутосомно-доминантные фенотипы». Менделевское наследование у человека: Каталоги аутосомно-доминантных, аутосомно-рецессивных и X-сцепленных фенотипов . Джонс Хопкинс Пресс. стр. 3–5.
- ^ «Запись — #123500 — СИНДРОМ КРУЗОНА — ОМИМ» . omim.org . Проверено 5 ноября 2022 г.
- ^ «Запись — #101400 — СИНДРОМ САЕТРЕ-ЧОТЦЕНА; SCS — OMIM» . omim.org . Проверено 5 ноября 2022 г.
- ^ Рирдон, В.; Зима, РМ (1994). «Синдром Сэтре-Чотцена» . Журнал медицинской генетики . 31 (5): 393–396. дои : 10.1136/jmg.31.5.393 . ISSN 0022-2593 . ПМЦ 1049872 . ПМИД 8064818 .
- ^ «Запись № 201000 — Синдром Карпентера 1; CRPT1» . omim.org . Проверено 4 декабря 2023 г.
- ^ Перейти обратно: а б «Запись 201020 — Акроцефалополисиндактилия IV типа» . omim.org . Проверено 4 декабря 2023 г.
- ^ «Запись 272350 — Синдром Саммитта» . omim.org . Проверено 4 декабря 2023 г.
- ^ «Запись 101120 — Акроцефалополисиндактилия III типа» . omim.org . Проверено 4 декабря 2023 г.
- ^ Перейти обратно: а б Андерсон, Питер. «Заголовки черепно-лицевой поддержки» (PDF) . Архивировано из оригинала 30 июня 2012 года.
- ^ «Синдром Пфайфера» . НОРД (Национальная организация по редким заболеваниям) . Проверено 5 ноября 2022 г.
Внешние ссылки
[ редактировать ]- Акроцефалосиндактилия Национальной медицинской библиотеки США по медицинским предметным рубрикам (MeSH)