Синдром Нунан с множественными лентиго
| Синдром Нунан с множественными лентиго (НСМЛ) | |
|---|---|
| Другие имена | синдром LEOPARD, кардиокожный синдром, синдром Горлина II, синдром профузного лентигиноза, прогрессирующий кардиомиопатический лентигиноз, [ 1 ] : 550 Синдром Капуте-Римоэна-Кёнигсмарка-Эстерли-Ричардсона, синдром Мойнахана |
 | |
| Вид лица в три четверти, пациент первого поколения с небольшим прогнатизмом и низко посаженными ушами. | |
| Специальность | Медицинская генетика |
Синдром Нунан с множественными лентиго ( NSML ), который является частью группы, называемой синдромами пути Ras / MAPK , [ 2 ] является редким аутосомно-доминантным заболеванием , [ 3 ] мультисистемное заболевание, вызванное мутацией в гене протеинтирозинфосфатазы , нерецепторного типа 11 ( PTPN11 ). Заболевание представляет собой комплекс признаков, в основном затрагивающих кожу, скелет и сердечно-сосудистую систему, которые могут присутствовать или не присутствовать у всех пациентов. Природа того, как мутация вызывает каждый из симптомов заболевания, малоизвестна; однако исследования продолжаются. Это РАСопатия .
Синдром Нунан с множественными лентиго вызван другой миссенс-мутацией того же гена. Синдром Нунан довольно распространен (от 1:1000 до 1:2500 живорождений), а также нейрофиброматоз 1 степени (который когда-то считался связанным с НМЛ) (1:3500); однако эпидемиологических данных по НМЛ не существует. [ 4 ]
Признаки и симптомы
[ редактировать ]Альтернативное название этого состояния, синдром LEOPARD, представляет собой мнемоническое слово , первоначально придуманное в 1969 году. [ 5 ] поскольку это состояние характеризуется некоторыми из следующих семи состояний, первые буквы которых обозначают ЛЕОПАРД, а также характерными « веснушками » кожи, вызванными лентиго , напоминающими большую кошку .
- Лентиго от красновато-коричневого до темно-коричневого цвета — пятна кожи (поверхностное поражение ), обычно встречающиеся в большом количестве (10 000+) на большой части кожи, иногда с покрытием более 80%. Они могут даже появиться внутри рта ( буккально ) или на поверхности глаза ( склерально ). Они имеют неровные границы и имеют размер от 1 мм в диаметре до пятен цвета кофе с молоком диаметром несколько сантиметров. Также участки витилигоподобной гипопигментации . некоторые могут наблюдаться
- электрокардиографической проводимости Нарушения : обычно наблюдаются на электрокардиографе в виде блокады пучка Гиса .
- Глазной гипертелоризм : широко расставленные глаза, что приводит к схожему сходству лиц у пациентов. Лицевые аномалии являются вторым по частоте симптомом после лентиго . К аномалиям также относятся: широкий корень носа, прогнатизм (выступающая нижняя челюсть) или низко посаженные, возможно повернутые уши.
- Легочный стеноз : Сужение легочной артерии на выходе из сердца . Могут присутствовать и другие сердечные аномалии, включая аортальный стеноз или пролапс митрального клапана .
- Аномальные гениталии : обычно крипторхизм (удержание яичек в теле) или монорхизм (одиночное яичко). У пациенток это проявляется отсутствием или одиночными яичниками, которые по своей природе гораздо труднее обнаружить. УЗИ проводится регулярно, начиная с возраста 1 года, для определения наличия яичников.
- Замедленный рост : медленный или задержка роста. Большинство новорожденных с этим синдромом имеют нормальный вес и рост при рождении, но в течение первого года их рост часто замедляется.
- Глухота : нейросенсорная (нервная глухота).
Наличие всех этих признаков не является необходимым для постановки диагноза. Клинический диагноз считается установленным, когда при наличии лентиго наблюдаются еще 2 симптома, такие как нарушения ЭКГ и глазной гипертелоризм, или при отсутствии лентиго наблюдаются 3 из вышеперечисленных состояний, при наличии родственника первой степени родства (т. е. родителя, ребенка, брат/сестра) с клиническим диагнозом. [ 6 ]
- Дополнительные дерматологические нарушения (подмышечные веснушки, локализованная гипопигментация , межпальцевые перепонки , гиперэластичная кожа)
- Легкая умственная отсталость наблюдается примерно у 30% больных синдромом.
- нистагм (непроизвольные движения глаз), судороги или гипосмия (снижение способности обоняния). У некоторых пациентов были зарегистрированы
- В 2004 году сообщалось о пациенте с рецидивирующей аневризмой верхних конечностей , потребовавшей хирургического вмешательства. [ 7 ]
- В 2006 году у пациента с НМЛ был зарегистрирован острый миелогенный лейкоз . [ 8 ]
Из-за редкости самого синдрома трудно определить, действительно ли некоторые дополнительные заболевания являются его частью. При базовой популяции, возможно, насчитывающей менее тысячи особей, один или два отдаленных случая могут очень быстро исказить статистическую совокупность.
-
 На руке 37-летнего пациента видна межпальцевая перепонка.
На руке 37-летнего пациента видна межпальцевая перепонка. -
 Пациент, 37 лет (второе поколение), гипертелоризм, широкий корень носа, небольшой птоз.
Пациент, 37 лет (второе поколение), гипертелоризм, широкий корень носа, небольшой птоз. -
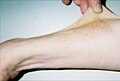 У тридцатисемилетнего пациента наблюдается гиперэластичность.
У тридцатисемилетнего пациента наблюдается гиперэластичность. -
 21-месячный пациент третьего поколения, подтвержденный генетическими тестами как Y279C , с глазным гипертелиоризмом, цефало-лицевым сходством.
21-месячный пациент третьего поколения, подтвержденный генетическими тестами как Y279C , с глазным гипертелиоризмом, цефало-лицевым сходством. -
 Туловище тридцатисемилетнего пациента во втором поколении с лентигинозом .
Туловище тридцатисемилетнего пациента во втором поколении с лентигинозом .





Патофизиология
[ редактировать ]
В двух преобладающих мутациях NSML ( Y279C и T468M ) мутации вызывают потерю каталитической активности белка SHP2 (генного продукта гена PTPN11 ), что является ранее нераспознанным поведением для этого класса мутаций. [ 9 ] Это мешает фактору роста и связанной с ним передаче сигналов. Хотя дальнейшие исследования подтверждают этот механизм, [ 10 ] [ 11 ] необходимы дополнительные исследования, чтобы определить, как это связано со всеми наблюдаемыми эффектами NSML.
Диагностика
[ редактировать ]Наличие заболевания можно подтвердить с помощью генетического теста. В исследовании 10 младенцев с клиническими признаками НМЛ до первого дня рождения у 8 (80%) пациентов было подтверждено подозрение на мутацию. еще одного пациента с подозрением на мутацию был обнаружен NF1 . Впоследствии после обследования матери у [ 12 ]
Идентифицировано 5 аллельных вариантов, ответственных за НСМЛ. Y279C , T468M , A461T , G464A и Q510P , которые, по-видимому, являются уникальной семейной мутацией, поскольку все остальные варианты вызваны ошибками перехода, а не трансверсией .
Уход
[ редактировать ]Рекомендуется, чтобы после постановки диагноза люди регулярно наблюдались у кардиолога, эндокринолога, дерматолога и других специалистов по мере появления симптомов.
Людям с синдромом, способным иметь детей, рекомендуется обратиться за генетической консультацией, прежде чем принять решение о рождении детей. Поскольку синдром часто проявляется как вариант forme fruste (неполная или необычная форма), необходимо провести обследование всех членов семьи. [ 13 ] Поскольку это аутосомно-доминантный признак, у каждого ребенка есть пятидесятипроцентная вероятность того, что он также родится с этим синдромом. Несмотря на полную пенетрантность, поскольку синдром имеет различную экспрессивность, у одного поколения синдром может быть легким, тогда как у следующего поколения он может быть сильно затронут.
После принятия решения о рождении детей и зачатия пары во время беременности за плодом наблюдают для оценки состояния сердца. При обнаружении грубого порока сердца родители получают консультацию по продолжению беременности.
Другое лечение представляет собой обычный уход при наличии симптомов: [ 13 ]
- Людям с эндокринными проблемами (низкий уровень тиреотропина [гормона гипофиза, отвечающего за регуляцию гормонов щитовидной железы], фолликулостимулирующего гормона ) рекомендуется медикаментозная терапия.
- Тем, кого беспокоит появление лентиго, может помочь криохирургия. Из-за большого количества лентиго это может занять много времени. Может помочь альтернативное лечение кремами с третиноином или гидрохиноном.
- Медикаментозная терапия для людей с сердечными нарушениями, поскольку эти нарушения становятся достаточно серьезными, чтобы оправдать использование этих методов лечения. ЭКГ обязательна перед любым хирургическим вмешательством из-за возможной аритмии .
Прогноз
[ редактировать ]Сам по себе НСМЛ не является опасным для жизни диагнозом, большинство людей с этим заболеванием живут нормальной жизнью. Обструктивная кардиомиопатия и другие патологические изменения, затрагивающие сердечно-сосудистую систему, могут быть причиной смерти у лиц с глубокими пороками сердца. [ 13 ]
Эпидемиология
[ редактировать ]В различной литературе этот синдром описывается как «редкий». [ 13 ] или «крайне редко». [ 14 ] Нет эпидемиологических данных о том, сколько людей страдают этим синдромом во всем мире; однако в медицинской литературе описано около 200 случаев. [ 15 ]
История
[ редактировать ]Zeisler и Becker впервые описали синдром с множественными лентиго , гипертелоризмом , pectus carinatum (выступанием грудины) и прогнатизмом (выступанием нижней челюсти) в 1936 году. [ 16 ] Спорадические описания добавлялись на протяжении многих лет. В 1962 году с этим заболеванием впервые были связаны нарушения сердечной деятельности и низкий рост. [ 17 ] В 1966 году добавились три семейных случая: мать, ее сын и дочь. [ 18 ] Еще один случай матери двух отдельных детей с разным отцовством двух детей был добавлен в 1968 году. [ 19 ]
Считалось, что еще в 2002 году [ 20 ] что синдром Нунан с множественными лентиго (НСМЛ) связан с нейрофиброматозом I типа (синдромом фон Реклингхаузена). Фактически, поскольку как в МКБ9, так и в МКБ10 отсутствует специальный диагностический код для НСМЛ, диагностический код для NF1 все еще иногда используется в диагностических целях, хотя было показано, что этот ген не связан с локусом NF1 . [ 21 ]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Джеймс, Уильям; Бергер, Тимоти; Элстон, Дирк (2005). Кожные заболевания Эндрюса: клиническая дерматология (10-е изд.). Сондерс. ISBN 0-7216-2921-0 .
- ^ Тидиман В.Е., Рауен К.А. (июнь 2009 г.). «РАСопатии: синдромы развития нарушения регуляции пути Ras/MAPK» . Текущее мнение в области генетики и развития . 19 (3): 230–6. дои : 10.1016/j.gde.2009.04.001 . ПМЦ 2743116 . ПМИД 19467855 .
- ^ Коппин Б.Д., Темпл И.К. (1997). «Синдром множественных лентиго (синдром LEOPARD или прогрессирующий кардиомиопатический лентигиноз)» . Журнал медицинской генетики . 34 (7): 582–6. дои : 10.1136/jmg.34.7.582 . ПМК 1051000 . ПМИД 9222968 .
- ^ Туллу М.С., Муранджан М.Н., Кантария В.К. и др. (1 апреля 2000 г.). «Синдром нейрофиброматоза-Нунана или синдром ЛЕОПАРДА? Клиническая дилемма» . J Постград Мед . 46 (2): 98–100. ПМИД 11013475 .
- ^ Горлин Р.Дж., Андерсон Р.К., Блоу М. (1969). «Синдром множественных лентигенов». Являюсь. Дж. Дис. Ребенок . 117 (6): 652–62. дои : 10.1001/archpedi.1969.02100030654006 . ПМИД 5771505 .
- ^ Ворон Д.А., Хэтфилд Х.Х., Калхофф Р.К. (1976). «Синдром множественных лентиго. Описание случая и обзор литературы». Являюсь. Дж. Мед . 60 (3): 447–56. дои : 10.1016/0002-9343(76)90764-6 . ПМИД 1258892 .
- ^ Ягубян М., Паннетон Дж.М., Линдор Н.М., Конти Э., Саркози А., Пиццути А. (апрель 2004 г.). «Синдром LEOPARD: новая ассоциация полианевризмы и обновленная информация о молекулярной генетике заболевания» . Дж. Васк. Сург . 39 (4): 897–900. дои : 10.1016/j.jvs.2003.11.030 . ПМИД 15071461 .
- ^ Учар С., Калискан У., Мартини С., Хейнритц В. (март 2006 г.). «Острый миеломоноцитарный лейкоз у мальчика с синдромом LEOPARD (положительная мутация гена PTPN11)». Ж. Педиатр. Гематол. Онкол . 28 (3): 123–5. дои : 10.1097/01.mph.0000199590.21797.0b . ПМИД 16679933 . S2CID 21559684 .
- ^ Тарталья М., Мартинелли С., Стелла Л. и др. (2006). «Разнообразие и функциональные последствия зародышевых и соматических мутаций PTPN11 при заболеваниях человека» . Американский журнал генетики человека . 78 (2): 279–90. дои : 10.1086/499925 . ПМК 1380235 . ПМИД 16358218 .
- ^ Ханна Н., Монтагнер А., Ли WH и др. (2006). «Снижение фосфатазной активности SHP-2 при синдроме LEOPARD: последствия связывания PI3K с Gab1». ФЭБС Летт . 580 (10): 2477–82. Бибкод : 2006FEBSL.580.2477H . doi : 10.1016/j.febslet.2006.03.088 . ПМИД 16638574 . S2CID 27676871 .
- ^ Контаридис М.И., Суонсон К.Д., Дэвид Ф.С., Барфорд Д., Нил Б.Г. (2006). «Мутации PTPN11 (Shp2) при синдроме LEOPARD имеют доминантно-негативные, а не активирующие эффекты» . Ж. Биол. Хим . 281 (10): 6785–92. дои : 10.1074/jbc.M513068200 . ПМИД 16377799 .
- ^ Диджилио MC, Саркози А, де Зорзи А и др. (2006). «Синдром LEOPARD: клинический диагноз на первом году жизни». Американский журнал медицинской генетики . 140 (7): 740–6. дои : 10.1002/ajmg.a.31156 . ПМИД 16523510 . S2CID 19570040 .
- ^ Перейти обратно: а б с д Синдром ЛЕОПАРДА в электронной медицине
- ^ «Синдром ЛЕОПАРДА» . НОРД — Национальная организация по редким заболеваниям.
- ^ «Синдром Нунан с множественными лентиго» . Национальная медицинская библиотека США.
- ^ Цейслер Э.П., Беккер С.В. (1936). «Генерализованное лентиго: его отношение к системным невыступающим невусам». Арка Дерматол Сифилол . 33 : 109–125. дои : 10.1001/archderm.1936.01470070112010 .
- ^ Мойнахан Э.Дж. (1962). «Множественные симметричные родинки с психическим и соматическим инфантилизмом и генитальной гипоплазией: первый мужской случай нового синдрома» . Труды Королевского медицинского общества . 55 (11): 959–960. дои : 10.1177/003591576205501112 . ПМК 1896920 . ПМИД 19994192 .
- ^ Вальтер Р.Дж., Полански Б.Дж., Гротис И.А. (1966). «Электрокардиографические нарушения в семье с генерализованным лентиго». Н. англ. Дж. Мед . 275 (22): 1220–5. дои : 10.1056/NEJM196612012752203 . ПМИД 5921856 .
- ^ Мэтьюз Н.Л. (1968). «Лентиго и электрокардиографические изменения». Н. англ. Дж. Мед . 278 (14): 780–1. дои : 10.1056/NEJM196804042781410 . ПМИД 5638719 .
- ^ Национальная медицинская библиотека MeSH: C05.660.207.525
- ^ Альбом Б.Е., Даль Н., Зеттерквист П., Аннерен Г. (1995). «Синдром Нунан с пятнами цвета кофе с молоком и синдром множественных лентиго не связаны с локусом нейрофиброматоза 1 типа». Клин. Жене . 48 (2): 85–9. дои : 10.1111/j.1399-0004.1995.tb04061.x . ПМИД 7586657 . S2CID 31291484 .
Внешние ссылки
[ редактировать ]- NSML в NIH / UW GeneTests
- Синдром Горлина II в журнале «Кто это назвал?»
- ДермАтлас 981603547
- Дермнетнц
- ДермИС