Генодерматоз
| Генодерматоз | |
|---|---|
| Другие имена | генодерматозы |
 | |
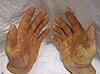
| Пациент с гидротической эктодермальной дисплазией Клаустона, одним из редких генодерматозов. | |
| Специальность | Дерматология , Медицинская генетика |
| Причины | Семейная история; генная мутация; хромосомная аномалия |
Генодерматоз — наследственное заболевание кожи с тремя наследственными типами, включая наследование одного гена , наследование множественных генов и хромосомное наследование. [ 1 ] Существует много различных типов генодерматоза; Распространенность генодерматоза колеблется от 1 на 6000 человек до 1 на 500 000 человек. [ 2 ] Генодерматоз оказывает влияние на консистенцию, цвет и структуру кутикулы кожи и соединительной ткани , конкретный участок поражения и клинические проявления на организме варьируют в зависимости от типа. [ 3 ] Несмотря на разнообразие и сложность генодерматоза, все же существуют некоторые распространенные методы, которые могут помочь людям диагностировать его. [ 4 ] После постановки диагноза разные типы генодерматоза требуют разных уровней терапии, включая вмешательства, сестринское вмешательство и лечение. [ 5 ] Среди них исследования терапии некоторых новых, сложных и редких типов все еще находятся на стадии разработки. [ 6 ] Воздействие генодерматоза можно наблюдать не только на теле, но и во всех аспектах жизни пациентов, включая, помимо прочего, психологическую, семейную жизнь, экономические условия и социальную деятельность. [ 5 ] [ 7 ] Соответственно, пациенты нуждаются в лечении, поддержке и помощи в этих областях. [ 5 ]
Наследственные режимы
[ редактировать ]Генодерматоз наследуется тремя способами: наследование одного гена, наследование множественных генов и наследование хромосом. [ 1 ]
Одиночный ген (моногенный)
[ редактировать ]Одногенное наследование генодерматоза относится к наследованию кожного заболевания, вызванного одной генетической аномалией, а наследственность одного гена делится в основном на четыре типа. [ 2 ]
Аутосомно-доминантное наследование
[ редактировать ]Первый вид – аутосомно-доминантное наследование, при этом типе наследования больные могут быть любого пола и их генодерматоз часто наследуется от одного из родителей. [ 2 ] К случаям кожных заболеваний, которые могут наследоваться по такому типу, относятся простой буллезный эпидермолиз (EBS), острая интермиттирующая порфирия , белый губчатый невус , ихтиоз , эпидермолитическая ладонно-подошвенная кератодермия, наследственный доброкачественный интраэпителиальный дискератоз и так далее. [ 1 ] [ 2 ] [ 8 ] [ 9 ]
Аутосомно-рецессивное наследование
[ редактировать ]Второй тип - аутосомно-рецессивное наследование, при этом типе наследования пациенты могут быть любого пола, и к такому наследованию обычно приводит инбридинг. [ 2 ] К случаям кожных заболеваний, которые могут наследоваться по такому типу, относятся буллезный эпидермолиз , пигментная ксеродермия , энтеропатический акродерматит , ихтиоз и так далее. [ 1 ] [ 2 ]
Х-сцепленное доминантное наследование
[ редактировать ]Третий вид – Х-сцепленное доминантное наследование, при этом виде наследования больные могут быть любого пола. [ 2 ] Пациенты мужского пола могут передать болезнь своим сыновьям, а шансы пациенток передать ее своим дочерям или сыновьям практически равны. [ 2 ] Случаи кожных заболеваний, которые могут наследоваться по такому типу, включают пигментное недержание мочи , очаговую гипоплазию дермы и т. д. [ 1 ] [ 8 ]
Х-сцепленное рецессивное наследование
[ редактировать ]Последний вид — Х-сцепленное рецессивное наследование, при этом виде наследования больные могут быть любого пола и распространенность у мужчин выше, чем у женщин. [ 2 ] Пациенты мужского пола не могут передать болезнь своим сыновьям. [ 2 ] К случаям кожных заболеваний, которые могут наследоваться по такому типу, относятся болезнь Фабри , ангидротическая эктодермальная дисплазия , врожденный дискератоз и так далее. [ 1 ] [ 2 ]
Множественный ген (полигенный)
[ редактировать ]Множественное наследование генодерматоза означает наследование кожного заболевания, вызванного множественными генетическими аномалиями. [ 2 ] Случаи кожных заболеваний, которые могут быть унаследованы по этому типу, включают витилиго , псориаз , обыкновенную пузырчатку , системную красную волчанку и так далее. [ 2 ] [ 8 ]
хромосома
[ редактировать ]Хромосомное наследование генодерматоза – это наследование кожного заболевания, вызванного хромосомной аномалией.
Одно и то же заболевание может наследоваться по-разному. Например, буллезный эпидермолиз может наследоваться по аутосомно-доминантному или аутосомно-рецессивному типу.
Типы
[ редактировать ]

Генодерматоз имеет множество типов, многие из которых встречаются редко.
Распространенные типы
[ редактировать ]Ихтиоз
[ редактировать ]Ихтиоз относится в основном к ихтиозу обыкновенному , распространенному генодерматозу, у людей с этим заболеванием наблюдается рыбная, сухая кожа, которая обычно появляется в раннем детстве и может исчезнуть в зрелом возрасте. [ 10 ] Распространенность вульгарного ихтиоза высока и поражает почти 1 из 250 человек. [ 11 ] Существуют также редкие виды ихтиоза, такие как эпидермолитический гиперкератоз, ихтиоз арлекина и так далее. [ 8 ]
Редкие виды
[ редактировать ]Синдром ребенка из шин Мишлен
[ редактировать ]Синдром ребенка шин Мишлен — редкий генодерматоз, возникающий при рождении, кожа пациентов уложена слоями симметрично, как изображение талисмана шины Мишлен , отсюда и название этого заболевания. [ 12 ]
Буллёзный эпидермолиз
[ редактировать ]Буллезный эпидермолиз — редкий тип генодерматоза, у людей с этим заболеванием на коже появляются волдыри, и это заболевание никогда полностью не излечивается на всю жизнь. [ 13 ] Буллезный эпидермолиз в основном подразделяют на четыре типа: дистрофический буллезный эпидермолиз , простой буллезный эпидермолиз, узловой буллезный эпидермолиз и синдром Киндлера . [ 13 ] Почти 1 из 50 000 человек страдает буллезным эпидермолизом. [ 14 ]
Врожденная пахионихия
[ редактировать ]


Врожденная пахионихия — редкий тип генодерматоза, клиническими проявлениями которого являются аномальное увеличение ногтей на руках и ногах, чрезмерное или слабое ороговение ладоней и подошв, повышенная потливость ладоней или подошв. [ 15 ] От 5000 до 10000 человек в мире страдают врожденной пахионихией. [ 16 ]
Эпидермолитическая ладонно-подошвенная кератодермия
[ редактировать ]Эпидермолитическая ладонно-подошвенная кератодермия часто появляется при рождении и полностью излечиться практически невозможно. [ 9 ] К клиническим проявлениям этого заболевания относятся чрезмерное ладонно-подошвенное ороговение, ладонные или подошвенные желтеют в разные стороны по краям или аномально повышенная потливость, а клинические проявления проявляются в симметричной форме на теле. [ 9 ]
Наследственный доброкачественный интраэпителиальный дискератоз
[ редактировать ]Наследственный доброкачественный интраэпителиальный дискератоз — редкий тип генодерматоза, который может возникнуть в младенчестве и раннем детстве, его симптомы часто проявляются в глазах и рту больных. [ 17 ] Глаза пациентов кажутся красными из-за расширения поверхностных сосудов и появления конъюнктивальных бляшек в глазах, во рту у пациентов могут быть толстые, мягкие и белые бляшки разного размера. [ 17 ] Весна – это острый приступ таких симптомов, как зуд, эритема, светобоязнь и так далее. [ 17 ]
Эпидермолитический гиперкератоз
[ редактировать ]Эпидермолитический гиперкератоз — редкий генодерматоз, который также называют нарушением ороговения 3-го типа и буллезной врожденной ихтиозиформной эритродермией, поражающий почти 1 на 200 000 — 300 000 человек. [ 18 ] Они также заявили, что его клинические проявления часто начинаются при рождении с крупных высыпаний по всему телу, а кожа пациентов настолько чувствительна, что даже легкие раны могут вызвать появление волдырей и шелушения. Потенциальные осложнения этого заболевания включают электролитные нарушения , сепсис и так далее. [ 18 ]
Гидротическая эктодермальная дисплазия
[ редактировать ]Гидротическая эктодермальная дисплазия — редкий генодерматоз, известный также как синдром Клустона. Ногти больных могут быть слишком толстыми, слишком ломкими, слишком изогнутыми или иметь разный цвет, волосы также появляются крапинками, редкими и другими аномалиями. [ 19 ] Эти симптомы часто начинаются, когда пациент является младенцем. [ 19 ]
Методы диагностики
[ редактировать ]Генодерматозы часто встречаются редко и разнообразно, однако методы диагностики имеют некоторые общие черты, диагностика редкого генодерматоза в основном делится на шесть этапов: 1. Каждый генодерматоз имеет различные клинические проявления. Люди могут наблюдать за особенностями и изменениями кожи, чтобы судить, больны они или нет. Характеристики будут разными в разных возрастных группах, поэтому необходимо наблюдать и фиксировать особенности кожи в каждой возрастной группе; 2. Генодерматоз – наследственное заболевание, поэтому знание как можно более детального и полного семейного анамнеза помогает при скрининге и диагностике; 3. Люди могут провести детальное физическое обследование, чтобы наблюдать особенности и проявления других органов, помимо кожи. Это может помочь сузить круг заболеваний и поставить окончательный диагноз; 4. Люди могут проводить лабораторные тесты, такие как биопсия кожи , с помощью высокотехнологичных и точных научных инструментов, чтобы получить дальнейшие результаты; 5. Различные генодерматозы и их клинические проявления могут быть обусловлены аномалиями одного и того же гена, а аномалии разных генов могут приводить к одним и тем же клиническим проявлениям. Чтобы поставить окончательный диагноз или выявить сложные и специфические типы генодерматоза, генотип – корреляция фенотипов требует внимания; 6. Если вышеуказанные пять шагов не помогли людям с подозрением на генодерматоз получить результат диагноза, им следует хранить всю свою информацию, такую как запись диагноза и клинические проявления на разных стадиях, и продолжать регистрировать изменения тело, ожидающее с позитивным настроем, медицина будущего может дать ответ. [ 4 ]
Терапия
[ редактировать ]Терапия генодерматоза требует не только заботы о коже больных, уменьшения их боли и предотвращения осложнений, но и оказания психологической поддержки больным и их семьям. [ 5 ]
Профилактика и уход
[ редактировать ]Различные виды генодерматоза требуют разных мер профилактики и ухода.
Ихтиоз
[ редактировать ]Радикального метода лечения ихтиоза не существует, но определенная осторожность может облегчить симптомы. [ 10 ] Чтобы предотвратить утолщение кожи и увлажнить ее, пациенты могут наносить крем, содержащий альфа-гидроксикислоты , а при последующих инфекциях пациентов также можно лечить антибиотиками. [ 10 ]
Буллёзный эпидермолиз
[ редактировать ]При буллезном эпидермолизе важен ежедневный уход. При обработке раны следите за ее чистотой и уменьшайте трение, используемые повязки и повязки должны быть нелипкими и мягкими, а пациент должен носить свободную одежду, чтобы не повредить рану. [ 13 ] [ 14 ]
Эпидермолитический гиперкератоз
[ редактировать ]Лечение эпидермолитического гиперкератоза направлено главным образом на контроль и облегчение симптомов, а хороший уход может снизить частоту осложнений, таких как электролитные нарушения и сепсис. [ 18 ] Чтобы улучшить внешний вид и самочувствие кожи, пациенты могут применять крем, содержащий альфа-гидроксикислоты, глицерин и мочевину , а при необходимости пациенты могут использовать антибиотики для борьбы с вторичными инфекциями. [ 18 ]
Врожденная пахионихия
[ редактировать ]Не существует радикального метода лечения врожденной пахионихии, но некоторая осторожность может облегчить симптомы. [ 16 ] Пациенты могут полировать и подстригать утолщенные ногти, некоторые из них могут использовать ретиноиды для облегчения симптомов, но это может усилить боль. [ 16 ]
Было показано, что селуметиниб и траметиниб уменьшают и контролируют рост опухоли у людей с нейрофиброматозом I типа, снижая вероятность злокачественных поражений. [ 20 ]
Терапевтические методы
[ редактировать ]Существуют новые и разрабатываемые генетические методы лечения генодерматоза. [ 20 ]
В случае Х-сцепленной гипогидротической эктодермальной дисплазии нерожденные дети с диагнозом этого заболевания могут лечиться еще в утробе матери. ребенка Обеспечение регуляторных белков в утробе матери в критический период роста ребенка может помочь скорректировать развитие потовых желез . [ 20 ]
Устекинумаб представляет собой биологический препарат, который можно использовать при различных генодерматозах, таких как врожденный ихтиоз, псориаз, дефицит антагониста рецептора интерлейкина-36 (ДИТРА) и так далее. [ 20 ]
Новый местный метод может лечить кожные аномалии при редких наследственных заболеваниях липидного обмена. [ 21 ] Этот метод препятствует аномальному мевалонату путем местного применения ловастатина , чтобы можно было максимально ингибировать выработку и кумуляцию ядовитых промежуточных продуктов метаболизма, и заменяет недостающий липид в коже путем местного применения холестерина . [ 20 ] [ 21 ] Идея о том, что аналогичные методы лечения могут быть разработаны и для других генодерматозов, также была высказана на ежегодной конференции Европейского общества дерматологов и венерологов. [ 21 ]
Что касается буллезного эпидермолиза, для лечения этого заболевания можно использовать метод под названием CRISPR путем анализа, модификации и замены генов, но этот метод является этически спорным, поскольку он допускает серьезное редактирование генов. [ 20 ]
Лечение некоторых редких заболеваний все еще изучается. Терапия генодерматоза требует обновления технологии, а развитие технологии зависит от постоянного понимания механизма заболевания. Исследования по лечению генодерматоза находятся на положительной стадии развития. [ 20 ]
Эффекты
[ редактировать ]Генодерматоз поражает пациентов по-разному. Генодерматоз — разновидность кожного заболевания, при котором поражается текстура, цвет и структура кутикулы и соединительной ткани кожи, некоторые из которых могут вызывать нарушения в работе других органов. [ 3 ] С социальной стороны, поскольку генодерматоз делает кожу и внешний вид пациентов отличными от обычных людей и ограничивает их в некоторых видах деятельности, они в той или иной степени сталкиваются с препятствиями в процессе приобретения друзей, поиска партнера, посещения школы и жизни. вход на рабочее место. [ 5 ] [ 7 ] Трудности в общении с другими людьми, а также мирские предрассудки могут повлиять на их психическое здоровье. Больные страдают от генодерматоза и в плане семейной жизни. [ 5 ] [ 7 ] Из-за поведенческих расстройств и лечения некоторых генодерматозов семьям приходится уделять больше времени уходу за пациентом, и у пациента может возникнуть больше беспокойства и размышлений о деторождении детей из-за этого заболевания. С экономической точки зрения лечение генодерматоза не является простым и коротким процессом, который создаст дополнительные семейные расходы и усилит экономическую нагрузку на больных и их семьи. [ 7 ]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Jump up to: а б с д и ж Бабу, Н. Аравиндха; Раджеш, Э.; Крупаа, Джаясри; Гнананандар, Г. (2015). «Генодерматозы» . Журнал фармации и биологических наук . 7 (Приложение 1): S203–S206. дои : 10.4103/0975-7406.155903 . ПМЦ 4439671 . ПМИД 26015711 .
- ^ Jump up to: а б с д и ж г час я дж к л м Филдс, Д. (июнь 2019 г.). Виды генодерматозов. Получено 8 сентября 2020 г. с https://www.news-medical.net/health/Types-of-Genodermatoses.aspx .
- ^ Jump up to: а б Баярт, Шерил Б.; Брандлинг-Беннетт, Хизер А. (2018). «Врожденные и наследственные заболевания кожи». Болезни Эйвери у новорожденных . стр. 1475–1494.e1. дои : 10.1016/B978-0-323-40139-5.00104-2 . ISBN 978-0-323-40139-5 .
- ^ Jump up to: а б Танчева-Пур, Илиана; Оджи, Винсент; Хас, Кристина (октябрь 2016 г.). «Многоэтапный подход к диагностике редких генодерматозов» . Журнал Немецкого дерматологического общества . 14 (10): 969–986. дои : 10.1111/ddg.13140 . ПМИД 27767270 . S2CID 46823821 .
- ^ Jump up to: а б с д и ж Фонд Рене Турена. (без даты). Генодерматозы и редкие кожные заболевания – приоритет общественного здравоохранения. Получено 8 сентября 2020 г. с https://www.frt-rareskin.org/Genodermatoses-Rare-Skin-Disorders .
- ^ Чамше, Жан Кристофер; Вуд, Гэри С.; Сиддики, Имтиаз А.; Сайед, Диба Н.; Адхами, Вакар М.; Тенг, Джойс М.; Мухтар, Хасан (июль 2012 г.). «Прогресс в области генетической и фармакологической терапии кератиновых генодерматозов: текущие перспективы и перспективы на будущее» . Экспериментальная дерматология . 21 (7): 481–489. дои : 10.1111/j.1600-0625.2012.01534.x . ПМК 3556927 . ПМИД 22716242 .
- ^ Jump up to: а б с д Дюфрен, Х.; Хадж-Рабия, С.; Бодемер, К. (ноябрь 2018 г.). «Влияние редкого хронического генодерматоза на повседневную жизнь семьи: на примере буллезного эпидермолиза». Британский журнал дерматологии . 179 (5): 1177–1178. дои : 10.1111/bjd.16710 . ПМИД 29710387 . S2CID 207077766 .
- ^ Jump up to: а б с д ДеСтефано, генеральный менеджер; Кристиано, AM (1 октября 2014 г.). «Генетика кожных заболеваний человека» . Перспективы Колд-Спринг-Харбора в медицине . 4 (10): а015172. doi : 10.1101/cshperspect.a015172 . ПМК 4200211 . ПМИД 25274756 .
- ^ Jump up to: а б с Чен, Нан; Сунь, Цзинъин; Сонг, Яли; Вэй, Синьцзин; Ши, Ян; Чжан, Ли (сентябрь 2017 г.). «Новая мутация гена KRT9 у китайского ханьца с эпидермолитической ладонно-подошвенной кератодермией». Журнал косметической дерматологии . 16 (3): 402–406. дои : 10.1111/jocd.12263 . ПМИД 27726289 . S2CID 23426593 .
- ^ Jump up to: а б с Национальный центр развития трансляционных наук. (2013). Ихтиоз обыкновенный. Получено с https://rarediseases.info.nih.gov/diseases/6752/ichthyosis-vulgaris .
- ^ Фонд ихтиоза и связанных с ним типов кожи. (без даты). Пресс-релиз: Исследование заболеваемости ихтиозом: Фонд ихтиоза и связанных с ним типов кожи (FIRST). Получено с http://www.firstskinfoundation.org/new-study-estimates-incidence-of-moderate-to-severe-ichthyosis.
- ^ Дхингра, Дхулика; Сетхи, Греция; Мантан, Мукта (август 2013 г.). «Малышка шин Мишлен: редкий генодерматоз» . Индийская педиатрия . 50 (8): 808. doi : 10.1007/s13312-013-0208-8 . ПМИД 24036654 . S2CID 38318738 .
- ^ Jump up to: а б с Правительство Квинсленда. (2017). Информационный бюллетень по буллёзному эпидермолизу. Получено 11 октября 2020 г. с https://www.childrens.health.qld.gov.au/fact-sheet-epidermolies-bullosa/ .
- ^ Jump up to: а б Национальная организация редких заболеваний (NORD). (2019). Буллёзный эпидермолиз. Получено с https://rarediseases.org/rare-diseases/epidermolies-bullosa/.
- ^ Агарвал, Пунит; Чхапервал, МахендраК; Сингх, Апурва; Верма, Арвинд; Ниджхаван, Маниша; Сингх, Кишор; Матур, Динеш (2013). «Врожденная пахионихия: редкий генодерматоз» . Индийский онлайн-журнал дерматологии . 4 (3): 225–7. дои : 10.4103/2229-5178.115527 . ПМЦ 3752484 . ПМИД 23984242 .
- ^ Jump up to: а б с Национальная организация редких заболеваний (NORD). (2018). Врожденная пахионихия. Получено из [1]
- ^ Jump up to: а б с О'Нил, MJ (17 мая 2013 г.). ДИСКЕРАТОЗ НАСЛЕДСТВЕННЫЙ ДОБРОКАЧЕСТВЕННЫЙ ИНТРАЭПИТЕЛИАЛЬНЫЙ; ХБИД. Получено из [2]
- ^ Jump up to: а б с д Квак, Джулиан; Маверакис, Эмануал (8 сентября 2006 г.). «Эпидермолитический гиперкератоз». Дерматологический онлайн-журнал . 12 (5): 6. дои : 10.5070/D38HV5R331 . ПМИД 16962021 . ПроКвест 68849936 .
- ^ Jump up to: а б Национальный центр развития трансляционных наук. (2020). Синдром Клоустона. Получено из [3]
- ^ Jump up to: а б с д и ж г Сильверберг, Нанетт (июль 2020 г.). «Новые методы лечения генодерматозов». Клиники по дерматологии . 38 (4): 462–466. doi : 10.1016/j.clindermatol.2020.03.006 . ПМИД 32972604 . S2CID 216334904 .
- ^ Jump up to: а б с Янсин, Брюс (16 ноября 2011 г.). «Местное лечение генодерматоза под названием «гениальность» » . Новости дерматологии .