Синдром затылочного рога
| Синдром затылочного рога | |
|---|---|
| Другие имена | синдром Элерса-Данлоса IX типа; X-Linked Кутис Лакса |
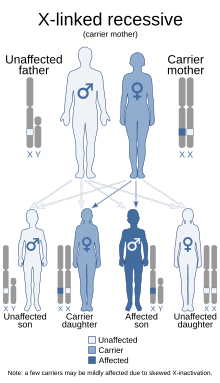 | |
| Это состояние наследуется по Х-сцепленному рецессивному типу. | |
| Специальность | Эндокринология |
| Осложнения | Аневризмы аорты |
| Медикамент | Дроксидопа , медь-гистидиновые инъекции |
Синдром затылочного рога ( СОЗ ), ранее считавшийся вариантом синдрома Элерса-Данлоса . [1] Х -сцепленное рецессивное заболевание митохондрий и соединительной ткани . Это вызвано дефицитом транспорта незаменимого минерала меди , связанным с мутациями гена ATP7A . [2] [3]
Считается, что только около 2/3 детей с СГЯ генетически унаследовали это заболевание; остальные 1/3 не имеют этого заболевания в семейном анамнезе. Поскольку заболевание является рецессивным, сцепленным с Х-хромосомой, оно поражает больше мужчин. Это связано с тем, что у них нет второй Х-хромосомы, в отличие от женщин, поэтому, по сути, у них отсутствует «резервная» копия с надлежащей функцией. Женщины гораздо чаще являются только носителями. Чтобы болезнь затронула женщину, она должна нести две дефектные Х-хромосомы, а не только одну. [4]
Заболевание считается более легким вариантом болезни Менкеса . [5]
Признаки и симптомы
[ редактировать ]Характеризуется дефицитом с желчью экскреции меди , что вызывает деформации скелета . К ним относятся выступы на задней части черепа ( парасагиттальные экзостозы костей , возникающие из затылочной кости — так называемые «затылочные рога»), а также деформации локтевого сустава , вывих головки лучевой кости , молоткообразные боковые концы ключиц и аномалии бедер и таза . [1] СГС проявляется в раннем и среднем детстве. [4] Дети могут представить такие особенности, как:
- Нормальный/немного замедленный интеллект
- Длинная шея, высокое арочное небо, длинное лицо, высокий лоб.
- Дряблость кожи и «двойной сустав».
- Паховые грыжи
- Перекручивание кровеносных сосудов
- Дивертикулы мочевого пузыря
- Дизавтономия — неспособность регулировать части нервной системы.
- Хроническая диарея
- Грубые волосы
- Низкий уровень церулоплазмина (9–29 мг/дл) [6]
- Поражение центральной нервной системы [7]
- Мышечная атрофия [7]
Причины
[ редактировать ]СГС представляет собой более легкий аллельный вариант болезни Менкеса, имеющий более поздний возраст начала и связанный с гораздо менее тяжелой центральной нейродегенерацией. Более мягкий характер OHS часто объясняется мутациями «протекающего» сплайсингового соединения, которые позволяют правильно процессировать 20–30% транскриптов информационной РНК (мРНК) ATP7A. Как и в случае болезни Менкеса, у людей с СГЯ наблюдаются аномалии соединительной ткани, возникающие в результате недостаточной активности лизилоксидазы, фермента, нуждающегося в меди, который обычно дезаминирует лизин и гидроксилизин на первом этапе образования поперечных связей коллагена. Такие люди также часто страдают от неприятных вегетативных признаков и симптомов, связанных с частичным дефицитом активности дофамин-β-гидроксилазы (ДБГ). DBH, еще один медь-зависимый фермент, обычно превращает дофамин в норадреналин, важнейший нейромедиатор в норадреналинэргических нейронах. Естественная мышиная модель СГС, так называемая пятнистая модель, воспроизводит аномалии соединительной ткани, дефицит DBH и легкие повреждения ЦНС, наблюдаемые у людей. [8]
Диагностика
[ редактировать ]Первоначальный диагноз болезни Менкеса (БМ) и ее более легких вариантов, таких как синдром затылочного рога, основан на клинических симптомах. Низкие уровни меди и церулоплазмина в сыворотке подтверждают клиническое подозрение на СГЯ, но необходимо биохимическое подтверждение с помощью культуры ткани. Окончательным диагностическим доказательством является демонстрация молекулярного дефекта в ATP7A. Наличие костных выступов на затылке подтверждает диагноз, и у некоторых пациентов их можно пальпировать. [9]
Уход
[ редактировать ]Курсы лечения детей с СГЯ зависят от тяжести случая. Дети с СГЯ часто получают физиотерапию и трудотерапию. [4] Им может потребоваться зонд для дополнительного питания, если они недостаточно растут. В попытке улучшить неврологическое состояние (судороги) в раннем возрасте ребенка можно сделать инъекции гистидина меди или хлорида меди.Однако инъекции гистидина меди оказались неэффективными для лечения соединительнотканных проявлений СГЯ. [10]
Прогноз
[ редактировать ]Долгосрочная естественная история СГС неизвестна. [8] Некоторые пациенты внезапно умерли в возрасте 17 лет. [11] тогда как один пациент дожил до 57 лет. [12] Причинами смерти являются дыхательная недостаточность, [11] аневризма аорты, [13] и внутричерепное кровоизлияние. [14]
Исследовать
[ редактировать ]NIH и Cyprium Therapeutics координируют совместную разработку генной терапии аденоассоциированного вируса под названием AAV-ATP7A для лечения болезни Менкеса и ее более легких вариантов, таких как синдром затылочного рога. [15] В марте 2017 года Cyprium Therapeutics приобрела всемирные права на разработку и коммерческие права на программу Menkes в NIH/NICHD через CRADA и лицензионные соглашения с NICHD. [16] AAV-ATP7A все еще находится на доклинической стадии, хотя FDA получило статус орфанного препарата. [ нужна ссылка ]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Jump up to: а б Интернет-менделевское наследование у человека (OMIM): 304150
- ^ Шайбер, Иво; Дринген, Ральф; Мерсер, Джулиан ФБ (2013). «Глава 11. Медь: последствия дефицита и перегрузки». В Астрид Сигель, Хельмут Сигель и Роланд К.О. Сигел (ред.). Взаимосвязь между ионами незаменимых металлов и заболеваниями человека . Ионы металлов в науках о жизни. Том. 13. Спрингер. стр. 359–387. дои : 10.1007/978-94-007-7500-8_11 . ISBN 978-94-007-7499-5 . ПМИД 24470097 .
- ^ Тан Дж., Робертсон С., Лем К.Э., Годвин С.С., Калер С.Г. (ноябрь 2006 г.). «Функциональный транспорт меди объясняет неврологическую сохранность синдрома затылочного рога» . Генетика в медицине . 8 (11): 711–8. дои : 10.1097/01.gim.0000245578.94312.1e . ПМИД 17108763 .
- ^ Jump up to: а б с Синдром Хорна. Архивировано 11 октября 2016 г. в Wayback Machine , 9 августа 2004 г.
- ^ Кеннерсон М.Л., Николсон Г.А., Калер С.Г., Ковальски Б., Мерсер Дж.Ф., Тан Дж. и др. (март 2010 г.). «Миссенс-мутации в гене переносчика меди ATP7A вызывают Х-сцепленную дистальную наследственную моторную нейропатию» . Американский журнал генетики человека . 86 (3): 343–52. дои : 10.1016/j.ajhg.2010.01.027 . ПМЦ 2833394 . ПМИД 20170900 .
- ^ Кодама Х., Мурата Ю., Кобаяши М. (август 1999 г.). «Клинические проявления и лечение болезни Менкеса и ее вариантов». Международная педиатрия . 41 (4): 423–9. дои : 10.1046/j.1442-200x.1999.01095.x . ПМИД 10453199 . S2CID 21267747 .
- ^ Jump up to: а б Вакаи С., Исикава Ю., Нагаока М., Окабе М., Минами Р., Хаякава Т. (май 1993 г.). «Поражение центральной нервной системы и генерализованная мышечная атрофия при синдроме затылочного рога: тип IX Элерса-Данлоса. Первый случай в Японии». Журнал неврологических наук . 116 (1): 1–5. дои : 10.1016/0022-510x(93)90081-9 . ПМИД 8099605 . S2CID 40538663 .
- ^ Jump up to: а б Калер С.Г. (январь 2011 г.). «Новые концепции и будущие тенденции заболеваний, связанных с переносом меди, связанных с ATP7A» . Обзоры природы. Неврология . 7 (1): 15–29. дои : 10.1038/nrneurol.2010.180 . ПМК 4214867 . ПМИД 21221114 .
- ^ Хорн Н., Тюмер З. (2003). «Глава 14: Болезнь Менкеса и синдром затылочного рога». В Royce PM, Steinmann B (ред.). Соединительная ткань и ее наследственные заболевания: молекулярные, генетические и медицинские аспекты (второе изд.). Уайли-Блэквелл. стр. 651–685. дои : 10.1002/0471221929.ch14 . ISBN 978-0-471-25185-9 .
- ^ Кодама Х., Фудзисава С., Бхадхпрасит В. (март 2011 г.). «Патология, клинические особенности и лечение врожденных нарушений обмена меди - основное внимание уделяется неврологическим аспектам». Мозг и развитие . 33 (3): 243–51. дои : 10.1016/j.braindev.2010.10.021 . ПМИД 21112168 . S2CID 38032382 .
- ^ Jump up to: а б Ясмин С., Лунд К., Де Паепе А., Де Би С., Хейберг А., Силва Дж., Мартинс М., Скьёрринге Т., Мёллер Л.Б. (апрель 2014 г.). «Синдром затылочного рога и классический синдром Менкеса, вызванные глубокими интронными мутациями, приводящими к активации псевдоэкзона ATP7A» . Европейский журнал генетики человека . 22 (4): 517–21. дои : 10.1038/ejhg.2013.191 . ПМЦ 3953917 . ПМИД 24002164 .
- ^ Бонати М.Т., Верде Ф., Хладник У., Каттелан П., Кампана Л., Кастроново С., Тикоцци Н., Мадерна Л., Коломбрита С., Папа С., Банфи П., Силани В. (декабрь 2017 г.). «Патогенный вариант ATP7A в семье, демонстрирующей вариабельный фенотип синдрома затылочного рога» . Отчеты о молекулярной генетике и метаболизме . 13 : 14–17. дои : 10.1016/j.ymgmr.2017.07.007 . ПМЦ 5522958 . ПМИД 28761814 .
- ^ Кирога Э., Хенеган Р. (июнь 2015 г.). «Аневризма брюшной аорты у пациента с синдромом затылочного рога 2» . Журнал случаев сосудистой хирургии . 1 (2): 138–140. дои : 10.1016/j.jvsc.2015.03.012 . ПМК 6849971 . ПМИД 31725130 .
- ^ Прауд В.К., Масселл Х.Г., Калер С.Г., Янг Д.В., Перси А.К. (октябрь 1996 г.). «Отличительный вариант болезни Менкеса с затылочными рогами: описание естественного течения и клинического фенотипа». Американский журнал медицинской генетики . 65 (1): 44–51. doi : 10.1002/(SICI)1096-8628(19961002)65:1<44::AID-AJMG7>3.0.CO;2-Y . ПМИД 8914740 .
- ^ Донсанте А., Йи Л., Зерфас П.М., Бринстер Л.Р., Салливан П., Гольдштейн Д.С., Прохаска Дж., Сентено Дж.А., Рашинг Э., Калер С.Г. (декабрь 2011 г.). «Добавление гена ATP7A к сосудистому сплетению приводит к долгосрочному устранению летального дефекта транспорта меди на мышиной модели болезни Менкеса» . Молекулярная терапия . 19 (12): 2114–23. дои : 10.1038/mt.2011.143 . ПМЦ 3242653 . ПМИД 21878905 .
- ^ «Корпоративная презентация» (PDF) . Cyprium Therapeutics, Inc. Октябрь 2017 г.