АТФ7А
| АТФ7А | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Идентификаторы | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Псевдонимы | ATP7A , DSMAX, MK, MNK, SMAX3, АТФаза, переносящая медь альфа | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Внешние идентификаторы | Опустить : 300011 ; МГИ : 99400 ; Гомологен : 35 ; Генные карты : ATP7A ; ОМА : ATP7A – ортологи | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Викиданные | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||




ATP7A , также известный как белок Менкеса ( MNK транспортирующую медь ), представляет собой АТФазу P-типа, , которая использует энергию, возникающую в результате гидролиза АТФ, для транспортировки Cu(I) через клеточные мембраны. Белок ATP7A является трансмембранным белком и экспрессируется в кишечнике и всех тканях, кроме печени. В кишечнике ATP7A регулирует всасывание Cu(I) в организме человека путем транспортировки Cu(I) из тонкого кишечника в кровь. В других тканях ATP7A перемещается между аппаратом Гольджи и клеточной мембраной для поддержания надлежащей концентрации Cu(I) (поскольку в клетке нет свободного Cu(I), все ионы Cu(I) прочно связаны) в клетке и обеспечивает определенные ферменты Cu(I) (например, пептидил-α-монооксигеназу , тирозиназу и лизилоксидазу ). Х-сцепленное наследственное летальное генетическое заболевание гена ATP7A вызывает болезнь Менкеса — дефицит меди, приводящий к ранней детской смертности. [5]
Ген
[ редактировать ]Ген ATP7A расположен на длинном (q) плече Х-хромосомы на участке Xq21.1. Кодируемый белок ATP7A содержит 1500 аминокислот. [6] Обнаружено по меньшей мере 12 мутаций этого гена, вызывающих заболевания. [7] Мутации/добавления/удаления этого гена часто вызывают дефицит меди, что приводит к прогрессирующей нейродегенерации и смерти детей. [8]
Структура
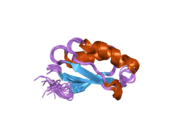
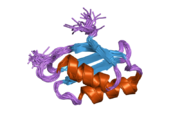
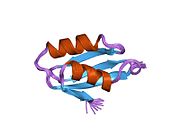
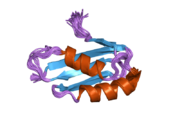
[ редактировать ]ATP7A представляет собой трансмембранный белок , N- и C-концы которого ориентированы к цитозолю (см. Рисунок). Он очень гомологичен белку ATP7B . ATP7A содержит три основных функциональных домена: [9] [10] [11] [12]
- Восемь трансмембранных сегментов , образующих канал и позволяющих Cu (I) проходить через мембрану;
- АТФ-связывающий домен;
- Большой N-концевой цитозольный домен, содержащий шесть повторяющихся сайтов связывания Cu(I), каждый из которых содержит мотив GMTCXXC.
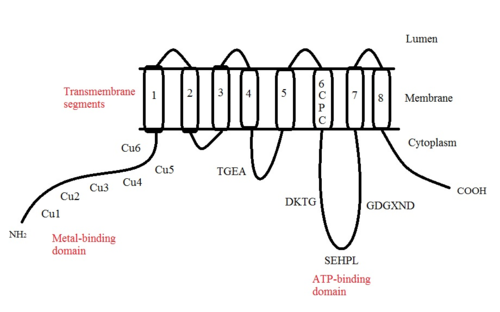
Многие мотивы в структуре ATP7A консервативны: [11]
- Мотив TGEA расположен в петле на цитозольной стороне между трансмембранными сегментами 4 и 5 и участвует в переносе энергии.
- Мотив CPC, расположенный в трансмембранном сегменте 6, является общим для всех АТФаз, транспортирующих тяжелые металлы.
Между трансмембранными сегментами 6 и 7 расположена большая цитоплазматическая петля, в которой расположены три мотива: DKTG, SEHPL и GDGXND.
- Мотив DKTG необходим для правильного функционирования АТФазы. Остаток аспарагиновой кислоты (D) фосфорилируется во время транспортных циклов.
- Мотив SEHPL существует только в АТФазах P-типа, транспортирующих тяжелые металлы. Без остатка гистидина (H) ATP7A может не функционировать должным образом.
- Считается, что мотив GDGXND вблизи трансмембранного сегмента 7 содержит в основном α-спирали и служит структурной опорой.
Шесть сайтов связывания Cu(I) на N-конце связывают по одному Cu(I) каждый. Этот сайт связывания неспецифичен для Cu(I) и может связывать ионы различных переходных металлов. Cd(II), Au(III) и Hg(II) связываются с местом связывания более прочно, чем Zn(II), тогда как Mn(II) и Ni(II) имеют более низкое сродство по сравнению с Zn(II). В случае Cu(I) возможный кооперативный механизм связывания наблюдается . Когда концентрация Cu(I) низкая, Cu(I) имеет более низкое сродство к ATP7A по сравнению с Zn(II); по мере увеличения концентрации Cu(I) наблюдается резкое увеличение сродства Cu(I) к белку. [11]
Конформационные изменения
[ редактировать ]Два остатка цистеина (C) в каждом сайте связывания Cu(I) координированы с Cu(I) с углом S-Cu(I)-S от 120 до 180° и расстоянием Cu-S 2,16 Å. Экспериментальные результаты с гомологичным белком ATP7B позволяют предположить, что в этом участвуют восстанавливающие реагенты, и при связывании Cu(I) дисульфидная связь между остатками цистеина разрывается, поскольку цистеин начинает связываться с Cu(I), что приводит к серии конформационных изменений на N-концевой белок и, возможно, активирующий Cu(I)-транспортирующую активность других цитозольных петель. [11]
Из шести центров связывания меди(I) два считаются достаточными для функции транспорта Cu(I). Причина существования шести сайтов связывания остается не до конца понятной. Однако некоторые ученые предположили, что остальные четыре места могут служить детекторами концентрации Cu(I). [9]
Транспортный механизм
[ редактировать ]АТФ7А принадлежит к семейству транспортеров, называемому АТФазами Р-типа , которые катализируют аутофосфорилирование ключевого консервативного остатка аспарагиновой кислоты (D) внутри фермента. Первым шагом является связывание АТФ с АТФ-связывающим доменом и связывание Cu(I) с трансмембранной областью. Затем ATP7A фосфорилируется по ключевому остатку аспарагиновой кислоты (D) в высококонсервативном мотиве DKTG, что сопровождается высвобождением Cu(I). Последующее дефосфорилирование промежуточного продукта завершает каталитический цикл. В каждом цикле ATP7A преобразуется как минимум в две разные конформации: E1 и E2. В состоянии E1 Cu(I) прочно связан с сайтами связывания на цитоплазматической стороне; в состоянии E2 сродство ATP7A к Cu(I) снижается и Cu(I) высвобождается на внеклеточной стороне. [13]
Функция
[ редактировать ]ATP7A важен для регуляции меди Cu(I) у млекопитающих. [10] Этот белок обнаруживается в большинстве тканей, но не экспрессируется в печени. [11] В тонком кишечнике белок ATP7A помогает контролировать всасывание Cu(I) из пищи. После того, как ионы Cu(I) абсорбируются энтероцитами , необходим ATP7A для их переноса через базолатеральную мембрану в кровоток. [9]
В других органах и тканях белок ATP7A выполняет двойную роль и перемещается между двумя местами внутри клетки. Белок обычно находится в клеточной структуре, называемой аппаратом Гольджи , которая модифицирует и транспортирует вновь вырабатываемые ферменты и другие белки. Здесь ATP7A поставляет Cu(I) определенным ферментам (например, пептидил-α-монооксигеназе , тирозиназе и лизилоксидазе). [9] ), которые имеют решающее значение для структур и функций мозга, костей, кожи, волос, соединительной ткани и нервной системы. Однако если уровни Cu(I) в клеточной среде повышены, ATP7A перемещается к клеточной мембране и удаляет избыток Cu(I) из клетки. [8] [10]
Функции АТФ7А в некоторых тканях организма человека следующие: [10]
| Салфетка | Расположение | Функция |
|---|---|---|
| Почка | Экспрессируется в эпителиальных клетках проксимальных и дистальных почечных канальцев. | Удаляет избыток Cu(I) для поддержания уровня Cu(I) в почках. |
| Паренхима | В цитотрофобласте , синцитиотрофобласте и эндотелиальных клетках сосудов плода. | Доставляет Cu(I) к плацентарным купроферментам и транспортирует Cu(I) в кровообращение плода. |
| Центральная нервная система | Различные локации | Распределяет Cu(I) в различных отделах центральной нервной системы. |
Взаимодействия
[ редактировать ]Было показано, что ATP7A взаимодействует с ATOX1 и GLRX . Антиоксидант 1 медный шаперон (ATOX1) необходим для поддержания гомеостаза меди Cu (I) в клетке. Он может связывать и транспортировать цитозольный Cu(I) к ATP7A в транс-сети Гольджи. Глутаредоксин-1 (GRX1) также необходим для функции ATP7A. Он способствует связыванию Cu(I) для последующего транспорта, катализируя восстановление дисульфидных мостиков. Он также может катализировать реакцию деглутатионилирования остатков C (цистеина) в шести Cu(I)-связывающих мотивах GMTCXXC. [10]
Клиническое значение
[ редактировать ]Болезнь Менкеса вызвана мутациями гена ATP7A. [14] Исследователи выявили различные мутации ATP7A, которые вызывают болезнь Менкеса и синдром затылочного рога (СГЗ), более легкую форму болезни Менкеса. Многие из этих мутаций удаляют часть гена и, по прогнозам, приводят к образованию укороченного белка ATP7A, который не способен транспортировать Cu(I). Другие мутации вводят дополнительные пары оснований ДНК или используют неправильные пары оснований, что приводит к тому, что белки ATP7A не функционируют должным образом. [6]
Измененные белки, возникающие в результате мутаций ATP7A, ухудшают усвоение меди из пищи, не могут доставлять медь определенным ферментам или застревают в клеточной мембране, не имея возможности перемещаться вперед и назад от аппарата Гольджи. В результате нарушения активности белка АТФ7А медь плохо распределяется по клеткам организма. Медь накапливается в некоторых тканях, таких как тонкий кишечник и почки, в то время как в мозге и других тканях уровень ее содержания необычно низок. [8] [9] Снижение поступления меди может снизить активность многочисленных медьсодержащих ферментов, которые необходимы для структуры и функционирования костей, кожи, волос, кровеносных сосудов и нервной системы. [8] [10] Медь также имеет решающее значение для распространения прионных белков, а у мышей с мутациями в Atp7a наблюдается отсроченное начало прионной болезни. [15] Стал доступен обширный ресурс клинически аннотированных генетических вариантов гена ATP7A. [16] подтверждение рекомендаций Американского колледжа медицинской генетики и геномики по интерпретации вариантов последовательностей.
Торможение
[ редактировать ]Было показано, что ингибитор протонной помпы омепразол блокирует АТФ7А в дополнение к его более известной роли по блокированию АТФ4А.
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Jump up to: а б с GRCh38: Версия Ensembl 89: ENSG00000165240 – Ensembl , май 2017 г.
- ^ Jump up to: а б с GRCm38: выпуск Ensembl 89: ENSMUSG00000033792 – Ensembl , май 2017 г.
- ^ «Ссылка на Human PubMed:» . Национальный центр биотехнологической информации, Национальная медицинская библиотека США .
- ^ «Ссылка на Mouse PubMed:» . Национальный центр биотехнологической информации, Национальная медицинская библиотека США .
- ^ Тюмер З., Мёллер Л.Б., Хорн Н. (1999). «Спектр мутаций ATP7A, дефектного гена при болезни Менкеса». Транспорт меди и его нарушения . Достижения экспериментальной медицины и биологии. Том. 448. стр. 83–95. дои : 10.1007/978-1-4615-4859-1_7 . ISBN 978-1-4613-7204-2 . ПМИД 10079817 .
- ^ Jump up to: а б Кодама Х., Мурата Ю. (август 1999 г.). «Молекулярная генетика и патофизиология болезни Менкеса». Международная педиатрия . 41 (4): 430–5. дои : 10.1046/j.1442-200x.1999.01091.x . ПМИД 10453200 . S2CID 19509148 .
- ^ Шимчикова Д., Хенеберг П. (декабрь 2019 г.). «Уточнение прогнозов эволюционной медицины на основе клинических данных о проявлениях менделевских болезней» . Научные отчеты . 9 (1): 18577. Бибкод : 2019NatSR...918577S . дои : 10.1038/s41598-019-54976-4 . ПМК 6901466 . ПМИД 31819097 .
- ^ Jump up to: а б с д и Луценко С., Гупта А., Беркхед Дж.Л., Зузель В. (август 2008 г.). «Клеточная многозадачность: двойная роль человеческих Cu-АТФаз в доставке кофакторов и внутриклеточном балансе меди» . Архив биохимии и биофизики . 476 (1): 22–32. дои : 10.1016/j.abb.2008.05.005 . ПМК 2556376 . ПМИД 18534184 .
- ^ Jump up to: а б с д и Бертини И., Грей Х., Стифель Э., Валентайн Дж. (2006). Биологическая неорганическая химия: строение и реакционная способность . Саусалито, Калифорния: Университетские научные книги. ISBN 978-1-891389-43-6 .
- ^ Инеси Дж., Пиланкатта Р., Тадини-Буонинсеньи Ф. (октябрь 2014 г.). «Биохимическая характеристика медных АТФаз Р-типа» . Биохимический журнал . 463 (2): 167–76. дои : 10.1042/BJ20140741 . ПМЦ 4179477 . ПМИД 25242165 .
- ^ Банки Л., Бертини И., Кантини Ф., Чиофи-Баффони С. (август 2010 г.). «Клеточное распределение меди: подход механистической системной биологии» . Клеточные и молекулярные науки о жизни . 67 (15): 2563–89. дои : 10.1007/s00018-010-0330-x . ПМЦ 11115773 . ПМИД 20333435 . S2CID 41967295 .
- ^ Хордыевска А., Попиолек Л., Кокот Ю. (август 2014 г.). «Многоликость меди в медицине и лечении» . Биометаллы . 27 (4): 611–21. дои : 10.1007/s10534-014-9736-5 . ПМЦ 4113679 . ПМИД 24748564 .
- ^ Сиггс О.М., Cruite JT, Du X, Ручманн С., Маслия Э., Бетлер Б., Олдстоун М.Б. (август 2012 г.). «Нарушение гомеостаза меди из-за мутации Atp7a задерживает начало прионной болезни» . Учеб. Натл. акад. наук. США . 109 (34): 13733–8. дои : 10.1073/pnas.1211499109 . ПМК 3427069 . ПМИД 22869751 .
- ^ «ATP7Agen - комплексный ресурс клинически аннотированных вариантов гена ATP7A» . clingen.igib.res.in . Проверено 6 июля 2020 г.
Дальнейшее чтение
[ редактировать ]- Барнс Н., Цивковский Р., Цивковская Н., Луценко С. (2005). «АтФазы, транспортирующие медь, белки Менкеса и болезни Вильсона играют различную роль во взрослом и развивающемся мозжечке» . J Биол Хим . 280 (10): 9640–5. дои : 10.1074/jbc.M413840200 . ПМИД 15634671 .
- Гриноф М., Пасе Л., Воскобойник И., Петрис М.Дж., О'Брайен А.В., Камакарис Дж. (2004). «Сигналы, регулирующие транспортировку АТФазы P-типа, транслоцирующей медь Менкеса (MNK; ATP7A), в поляризованных клетках MDCK». Am J Physiol Cell Physiol . 287 (5): C1463–71. doi : 10.1152/ajpcell.00179.2004 . ПМИД 15269005 . S2CID 40168524 .
- Мёллер Л.Б., Тюмер З., Лунд С., Петерсен С., Коул Т., Хануш Р., Зайдель Дж., Йенсен Л.Р., Хорн Н. (2000). «Подобные мутации в сайте сплайсинга гена ATP7A приводят к различным фенотипам: классической болезни Менкеса или синдрому затылочного рога» . Ам Джей Хум Жене . 66 (4): 1211–20. дои : 10.1086/302857 . ПМЦ 1288188 . ПМИД 10739752 .
- Воскобойник И, Камакарис Дж (2002). «АТФаза Менкеса, транслоцирующая медь P-типа (ATP7A): биохимические и клеточные биологические свойства, а также роль в болезни Менкеса». J Bioenerg Biomembr . 34 (5): 363–71. дои : 10.1023/A:1021250003104 . ПМИД 12539963 . S2CID 23109512 .
- Харрис ЭД, Редди MC, Цянь Ю, Тиффани-Кастильони Э, Маджумдар С, Нельсон Дж (1999). «Множественные формы Cu-АТФазы Менкеса». Транспорт меди и его нарушения . Достижения экспериментальной медицины и биологии. Том. 448. стр. 39–51. дои : 10.1007/978-1-4615-4859-1_4 . ISBN 978-1-4613-7204-2 . ПМИД 10079814 .
- Кокс Д.В., Мур С.Д. (2003). «Медь, транспортирующая АТФазы P-типа, и болезни человека». Дж. Биоэнергетика. Биомембр . 34 (5): 333–8. дои : 10.1023/А:1021293818125 . ПМИД 12539960 . S2CID 21471699 .
- Воскобойник И, Камакарис Дж (2003). «АТФаза Менкеса, транслоцирующая медь P-типа (ATP7A): биохимические и клеточные биологические свойства, а также роль в болезни Менкеса». Дж. Биоэнергетика. Биомембр . 34 (5): 363–71. дои : 10.1023/A:1021250003104 . ПМИД 12539963 . S2CID 23109512 .
- Ла Фонтен С., Мерсер Дж. Ф. (2007). «Торговля медь-АТФазами, ATP7A и ATP7B: роль в гомеостазе меди». Арх. Биохим. Биофиз . 463 (2): 149–67. дои : 10.1016/j.abb.2007.04.021 . ПМИД 17531189 .
- Луценко С., ЛеШейн Е.С., Шинде У (2007). «Биохимические основы регуляции медьтранспортирующих АТФаз человека» . Арх. Биохим. Биофиз . 463 (2): 134–48. дои : 10.1016/j.abb.2007.04.013 . ПМК 2025638 . ПМИД 17562324 .
- Дирик Х.А., Амброзини Л., Спенсер Дж., Гловер Т.В., Мерсер Дж.Ф. (1996). «Молекулярная структура гена болезни Менкеса (ATP7A)». Геномика . 28 (3): 462–9. дои : 10.1006/geno.1995.1175 . ПМИД 7490081 .
- Тюмер З., Вурал Б., Тоннесен Т., Челли Дж., Монако А.П., Хорн Н. (1995). «Характеристика экзонной структуры гена болезни Менкеса с использованием векторной ПЦР». Геномика . 26 (3): 437–42. дои : 10.1016/0888-7543(95)80160-N . ПМИД 7607665 .
- Калер С.Г., Галло Л.К., Прауд В.К., Перси А.К., Марк Ю., Сигал Н.А., Гольдштейн Д.С., Холмс К.С., Гал В.А. (1995). «Синдром затылочного рога и легкий фенотип Менкеса, связанный с мутациями сайта сплайсинга в локусе MNK». Нат. Жене . 8 (2): 195–202. дои : 10.1038/ng1094-195 . ПМИД 7842019 . S2CID 12122103 .
- Дас С., Левинсон Б., Уитни С., Вулпе С., Пакман С., Гитшир Дж. (1994). «Различные мутации у пациентов с болезнью Менкеса часто приводят к пропуску экзонов» . Являюсь. Дж. Хум. Жене . 55 (5): 883–9. ЧВК 1918324 . ПМИД 7977350 .
- Челли Дж., Тюмер З., Тоннесен Т., Петтерсон А., Исикава-Бруш Ю., Томмерап Н., Хорн Н., Монако АП (1993). «Выделение гена-кандидата болезни Менкеса, который кодирует потенциальный белок, связывающий тяжелые металлы». Нат. Жене . 3 (1): 14–9. дои : 10.1038/ng0193-14 . ПМИД 8490646 . S2CID 205341350 .
- Мерсер Дж. Ф., Ливингстон Дж., Холл Б., Пейнтер Дж. А., Беги С., Чандрасекхараппа С., Локхарт П., Граймс А., Бхаве М., Семениак Д. (1993). «Выделение частичного гена-кандидата болезни Менкеса путем позиционного клонирования». Нат. Жене . 3 (1): 20–5. дои : 10.1038/ng0193-20 . ПМИД 8490647 . S2CID 9148871 .
- Вулпе С., Левинсон Б., Уитни С., Пакман С., Гитшир Дж. (1993). «Выделение гена-кандидата болезни Менкеса и доказательства того, что он кодирует АТФазу, переносящую медь». Нат. Жене . 3 (1): 7–13. дои : 10.1038/ng0193-7 . ПМИД 8490659 . S2CID 24883244 .
- Левинсон Б., Конант Р., Шнур Р., Дас С., Пакман С., Гитшир Дж. (1997). «Повторяющийся элемент в регуляторной области гена MNK и его делеция у пациента с синдромом затылочного рога». Хм. Мол. Жене . 5 (11): 1737–42. дои : 10.1093/hmg/5.11.1737 . ПМИД 8923001 .
- Ямагучи Ю., Хейни М.Э., Сузуки М., Гитлин Дж.Д. (1997). «Биохимическая характеристика и внутриклеточная локализация белка болезни Менкеса» . Учеб. Натл. акад. наук. США . 93 (24): 14030–5. дои : 10.1073/pnas.93.24.14030 . ЧВК 19489 . ПМИД 8943055 .
- Петрис М.Дж., Мерсер Дж.Ф., Калвенор Дж.Г., Локхарт П., Глисон П.А., Камакарис Дж. (1997). «Лиганд-регулируемый транспорт откачивающего насоса медной АТФазы Менкеса P-типа из аппарата Гольджи к плазматической мембране: новый механизм регулируемого транспорта» . ЭМБО Дж . 15 (22): 6084–95. дои : 10.1002/j.1460-2075.1996.tb00997.x . ПМК 452430 . ПМИД 8947031 .
- Тюмер З., Лунд С., Толшав Дж., Вурал Б., Тоннесен Т., Хорн Н. (1997). «Идентификация точковых мутаций у 41 неродственного пациента, страдающего болезнью Менкеса» . Являюсь. Дж. Хум. Жене . 60 (1): 63–71. ПМЦ 1712537 . ПМИД 8981948 .
- Дирик Х.А., Адам А.Н., Эскара-Уилке Дж.Ф., Гловер Т.В. (1997). «Иммуноцитохимическая локализация белка-транспортера меди Менкеса (ATP7A) в транс-сети Гольджи» . Хм. Мол. Жене . 6 (3): 409–16. дои : 10.1093/hmg/6.3.409 . ПМЦ 7185191 . ПМИД 9147644 .
- Ронс Н., Муарард, член парламента, Робб Л., Тутэн А., Виллард Л., Морейн С. (1997). «Переход C2055T в экзоне 8 гена ATP7A связан с пропуском экзона в семействе синдромов затылочного рога» . Являюсь. Дж. Хум. Жене . 61 (1): 233–8. дои : 10.1016/S0002-9297(07)64297-9 . ПМК 1715861 . ПМИД 9246006 .
- Гитшир Дж., Моффат Б., Рейли Д., Вуд В.И., Фэйрбразер У.Дж. (1998). «Структура раствора четвертого металлосвязывающего домена АТФазы Менкеса, транспортирующей медь». Нат. Структура. Биол . 5 (1): 47–54. дои : 10.1038/nsb0198-47 . ПМИД 9437429 . S2CID 172550 .
Внешние ссылки
[ редактировать ]- ATP7A+белок+человек Национальной медицинской библиотеки США по медицинским предметным рубрикам (MeSH)
- Запись GeneReviews/NCBI/NIH/UW о связанных с ATP7A нарушениях транспорта меди, включая: болезнь Менкеса, синдром затылочного рога, дистальную моторную невропатию, связанную с ATP7A.
- Записи OMIM о нарушениях транспорта меди, связанных с ATP7A
- GeneCard
- человека Местоположение генома ATP7A и страница сведений о гене ATP7A в браузере генома UCSC .
галерея PDB |
|---|