Синдром крика
| Синдром крика | |
|---|---|
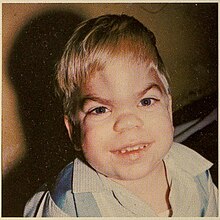 | |
| Пациент с синдромом Херлера | |
| Причины | Дефицит фермента альфа-L идиронидазы |
| Дифференциальный диагноз | Синдром Херлера-Шей ; Синдром Шей ; Синдром Охотника ; Другие мукополисахаридозы |
| Прогноз | Смерть обычно происходит до 12 лет |
| Частота | 1 из 100 000 |
Синдром Херлера , также известный как мукополисахаридоз типа IH ( MPS-IH ), болезнь Херлера и ранее Горгоилизм , является генетическим заболеванием , которое приводит к накоплению больших молекул сахара, называемых гликозаминогликанами (GAGS) в лизосом . Неспособность разрушить эти молекулы приводит к широкому разнообразию симптомов, вызванных повреждением нескольких различных систем органов , включая, помимо прочего, нервную систему , скелетную систему , глаза и сердце .
Основным механизмом является дефицит альфа-L идиронидазы , фермента, ответственного за разрушение кляп. [ 1 ] : 544 наращивание сульфата дерматана и гепаран Без этого фермента в организме происходит . Симптомы появляются в детстве, и обычно происходит ранняя смерть. Другие, менее тяжелые формы MPS типа I включают синдром Hurler-Scheie (MPS-IHS) и синдром Шей (MPS-IS).
Синдром Херлера классифицируется как лизосомальная болезнь хранения . Он клинически связан с синдромом Хантера (MPS II); [ 2 ] Тем не менее, синдром Охотника связан с X , а синдром Херлера аутосомно-рецессивный .
Признаки и симптомы
[ редактировать ]
У детей с синдромом Херлера может показаться нормальным при рождении и развиваться в течение первых лет жизни. Симптомы варьируются между пациентами. [ Цитация необходима ]
Одним из первых нарушений, которые могут быть обнаружены, является скорлупом особенности лица; Эти симптомы могут начаться в возрасте 3–6 месяцев. Голова может быть большой с выдающимися лобными костями . Череп может быть удлинен . Нос может иметь сплюснутый носовый мост с непрерывными носовыми выделениями. Глазки могут быть широко расставлены, и глаза могут выступать из черепа. Губы могут быть большими, и пострадавшие дети могут постоянно удерживать челюсти. Аномалии скелета происходят примерно к 6 месяцам, но могут быть клинически очевидными до 10–14 месяцев. Пациенты могут испытывать изнурительные деформации позвоночника и тазобедренного сустава, синдром запястного канала и жесткость сустава. Пациенты могут иметь нормальную высоту в младенчестве, но перестать расти в возрасте до двух лет. Они не могут достигать высоты более 4 футов (1,2 м). [ Цитация необходима ]
Другие ранние симптомы могут включать паховые и пупочные грыжи . Они могут присутствовать при рождении, или они могут развиваться в течение первых месяцев жизни. Заталевание роговицы и дегенерации сетчатки могут возникнуть в течение первого года жизни, что приведет к слепоте. Увеличенная печень и селезенка распространены. Не существует дисфункции органов, но осаждение GAG в этих органах может привести к огромному увеличению размера. Пациенты также могут иметь диарею . аортального клапана . Может возникнуть болезнь [ Цитация необходима ]
Обструкция дыхательных путей является частой, обычно вторичной по отношению к аномальным шейным позвонкам. [ 3 ] Инфекции верхних и нижних дыхательных путей могут быть частыми. [ Цитация необходима ]
Задержка развития может стать очевидной к возрасту 1–2 года, с максимальным функциональным возрастом 2–4 года. Прогрессивное ухудшение следует. Большинство детей разрабатывают ограниченные языковые возможности. Смерть обычно происходит к 10 годам. [ 4 ] [ 5 ]
Генетика
[ редактировать ]
Дети с синдромом Херлера несут две дефектные копии гена Идуа , которые были сопоставлены с участком 4P16,3 на хромосоме 4 . Это ген, который кодирует для белка идоронидазы. По состоянию на 2018 год [update] , что более 201 различных мутаций в гене ИДУА вызывают депутаты I. Было показано [ 6 ]
Поскольку синдром Херлера является аутосомно -рецессивным расстройством, пораженные люди имеют две неработающие копии гена. Человек, родившийся с одной обычной копией и одной дефектной копией, называется перевозчиком . Они будут продуцировать меньше α-L-идуронидазы, чем индивид с двумя нормальными копиями гена. Однако снижение выработки фермента у носителей остается достаточным для нормальной функции; Человек не должен проявлять никаких симптомов заболевания. [ Цитация необходима ]
Механизмы
[ редактировать ]
Ген Idua отвечает за кодирование фермента, называемого альфа-L-идуронидазой. Благодаря гидролизу альфа-L-идуронидаза отвечает за разрушение молекулы, называемой унсульфатированной альфа-L-идуроновой кислотой . Это мороновая кислота , содержащаяся в сульфате дерматана и гепаран -сульфат. Фермент альфа-L-идуронидазы расположен в лизосомах. Без достаточной ферментативной функции эти приколы не могут быть усваиваются должным образом. [ 7 ]
Диагноз
[ редактировать ]Диагноз часто может быть получен с помощью клинического обследования и тестов мочи (избыточные мукополисахариды выделяются в моче ). Анализы ферментов (тестирование различных клеток или жидкостей тела в культуре на дефицит ферментов) также используются для обеспечения окончательной диагностики одного из мукополисахаридозов. Пренатальный диагноз с использованием амниоцентеза и хорионной выборки ворсинки может проверить, несет ли плод либо копию дефектного гена , либо подвержен влиянию расстройства. Генетическое консультирование может помочь родителям, у которых есть семейная история мукополисахаридозов, определить, несут ли они мутированный ген, который вызывает расстройства. [ Цитация необходима ]
Классификация
[ редактировать ]Все члены семейства мукополисахаридоза также являются лизосомальными заболеваниями хранения . Мукополисахаридоз типа I (MPS I) делится на три подтипа на основе тяжести симптомов. Все три типа приводят к отсутствию или снижению функционирования одного и того же фермента. MPS-IH (синдром Херлера) является наиболее тяжелым из подтипов MPS I. Двумя другими типами являются MPS-IS ( синдром Шей ) и MPS-IHS ( синдром Hurler-Scheie ). [ Цитация необходима ]
Из -за существенного совпадения синдрома Херлера, синдрома Херлера -Шей и синдромом Шей, некоторые источники считают, что эти термины устарели. Вместо этого депутаты я могу быть разделен на «тяжелые» и «ослабленные» формы. [ 8 ]
Уход
[ редактировать ]В настоящее время нет лекарства от синдрома Херлера. Заместительная терапия ферментами с идуронидазой (альдуразим) может улучшить легочную функцию и подвижность. Это может уменьшить количество углеводов, которые неправильно хранятся в органах. Хирургическая коррекция деформаций рук и ног может быть необходимой. Хирургия роговицы может помочь смягчить проблемы со зрением. [ 5 ]
Трансплантация костного мозга (BMT) и трансплантация пуповинной крови (UCBT) можно использовать в качестве лечения MPS I. BMT от братьев и сестер с идентичными генами HLA и от родственников с аналогичными генами HLA может значительно улучшить выживаемость, когнитивную функцию и физические симптомы. У пациентов может развиться заболевание трансплантата по сравнению с болезнью хозяина ; Это, скорее всего, у не сестер доноров. В исследовании 1998 года у детей с HLA-идентичных донорами братьев и сестер была пятилетняя выживаемость 75%; Дети с не сестрами доноров имели пятилетнюю выживаемость в 53%. [ 9 ]
Дети часто не хватает доступа к подходящему донору костного мозга. В этих случаях UCBT от неродственных доноров может увеличить выживаемость, снизить физические признаки заболевания и улучшить познание. Осложнения от этого лечения могут включать в себя болезнь трансплантата по сравнению с хозяином . [ 10 ]
Прогноз
[ редактировать ]Британское исследование 2008 года показало среднюю предполагаемую продолжительность жизни в 8,7 года для пациентов с синдромом Херлера. Для сравнения, средняя продолжительность жизни для всех форм MPS типа I составила 11,6 года. Пациенты, которые получали успешную трансплантацию костного мозга, имели 2-летнюю выживаемость 68% и 10-летняя выживаемость 64%. Пациенты, которые не получали пересадки костного мозга, имели значительно сниженную продолжительность жизни, со средним возрастом 6,8 года. [ 4 ]
Эпидемиология
[ редактировать ]Синдром Херлера имеет общую частоту одного на 100 000. [ 5 ] В совокупности все мукополисахаридозы имеют частоту примерно каждую из каждых 25 000 рождений в Соединенных Штатах. [ 2 ]
Исследовать
[ редактировать ]Генная терапия
[ редактировать ]Большой интерес существует при лечении депутатов I с генной терапией . На животных моделях доставка гена идуронидазы была выполнена с помощью ретровируса , аденовируса , аденоассоциированного вируса и плазмидных векторов. Мыши и собаки с депутатами меня успешно лечили генной терапией. Большинство векторов могут исправить заболевание в печени и селезенке и могут исправить эффекты мозга с высокой дозой. Генная терапия улучшила выживаемость, неврологические и физические симптомы; Тем не менее, у некоторых животных развились необъяснимые опухоли печени. Если проблемы безопасности могут быть решены, генная терапия может обеспечить альтернативное лечение человека для расстройств MPS в будущем. [ 11 ]
Sangamo Therapeutics , штаб -квартира в Ричмонде, штат Калифорния , в настоящее время проводит клиническое исследование, включающее редактирование генов с использованием нуклеазы цинкового пальца (ZFN) для лечения MPS I. [ 12 ]
История
[ редактировать ]В 1919 году Гертруд Херлер , немецкий педиатр, описал синдром, включающий в себя орушение роговицы, аномалии скелета и умственную отсталость. Аналогичная болезнь «Горгоилизма» была описана в 1917 году Чарльзом А. Хантером. Херлер не упомянул газету Хантера. Из -за прерываний связи, вызванных Первой мировой войной , вполне вероятно, что она не знала о его исследовании. Синдром Херлера теперь относится к депутатам IH, в то время как синдром Охотника относится к MPS II. [ 13 ] [ 14 ] В 1962 году Scheie идентифицировал более мягкую форму депутатов, что привело к обозначению синдрома Шей. [ 4 ]
Смотрите также
[ редактировать ]- Синдром Охотника (MPS II)
- Синдром Санфилиппо (MPS III)
- Синдром Моркио (MPS IV)
- Мароты - Синдром Ламари (MPS VI)
Ссылки
[ редактировать ]- ^ Джеймс В.Д., Бергер Т.Г. и др. (2006). Болезни Эндрюса кожи: клиническая дерматология . Saunders Elsevier. ISBN 978-0-7216-2921-6 .
- ^ Jump up to: а беременный «Мукополисахаридозы фактической информационной информационной информационной бюллетеня» . Национальный институт неврологических расстройств и инсульта . 15 ноября 2017 года . Получено 11 мая 2018 года .
- ^ Myer CM (июль 1991 г.). «Обструкция дыхательных путей при синдроме Херлера-радиографические особенности». Международный журнал педиатрической оториноларингологии . 22 (1): 91–6. doi : 10.1016/0165-5876 (91) 90101-G . PMID 1917344 .
- ^ Jump up to: а беременный в Мур Д., Конгон М.Дж., Призрак Е., Лавери С (сентябрь 2008 г.). «Распространенность и выживание при мукополисахаридозе I: Hurler, Hurler-Scheie и Scheie Synddromes в Великобритании» . ОРФАНЕТНЫЙ ЖУРНАЛ РЕДИЧНЫХ БОЛЬШЕ . 3 : 24. DOI : 10.1186/1750-1172-3-24 . PMC 2553763 . PMID 18796143 .
- ^ Jump up to: а беременный в BANIKAZEMI M (12 октября 2014 г.). «Синдром Херлера, синдром Херлера-Шей и синдром Шей (мукополисахаридоз типа I)» . Medscape . Получено 10 мая 2018 года .
- ^ Chkioua L, Boudabous H, Jaballi I, Grissa O, Turkia HB, Tebib N, Laradi S (май 2018). «Новая мутация гена идуа в новой сплайке в родословных из тунисского синдрома с синдромом Херлера» . Диагностическая патология . 13 (1). Biomed Central : 35. DOI : 10.1186/S13000-018-0710-3 . PMC 5975427 . PMID 29843745 .
- ^ "Идуа ген" . Генетика дома ссылка. 11 июня 2019 года . Получено 18 июня 2019 года .
- ^ «Мукополисахаридоз типа I» . Генетика дома ссылка . Получено 10 мая 2018 года .
- ^ Peters C, Shapiro EG, Anderson J, Henslee-Downey PJ, Klemperer MR, Cowan MJ, et al. (Апрель 1998 г.). «Синдром Херлера: II. Результат HLA-генотипно-идентичной сестры и трансплантации доноров, связанных с HLA-HAPLOIDATICAL, у пятидесяти детей. Исследовательская группа совместной работы по болезни» . Кровь . 91 (7): 2601–8. doi : 10.1182/blood.v91.7.2601 . PMID 9516162 .
- ^ Стапса С.Л., Эсколер М.Л., По М., Ким Й., Мартин П.Л., С -Саболкс П. и др. (Май 2004 г.). «Трансплантация шнурной крови от неродственных доноров у пациентов с синдромом Херлера» . Новая Англия Журнал медицины . 350 (19): 1960–9. doi : 10.1056/nejmoa032613 . PMID 15128896 . S2CID 43572313 .
- ^ Ponder KP, Haskins Me (сентябрь 2007 г.). «Генная терапия при мукополисахаридозе» . Экспертное мнение о биологической терапии . 7 (9): 1333–45. doi : 10.1517/14712598.7.9.1333 . PMC 3340574 . PMID 17727324 .
- ^ «Восходящая доза-исследование редактирования генома с помощью нуклеазы цинкового пальца (ZFN) терапевтического SB-318 у субъектов с MPS I» . Clinicaltrials.gov . Национальная библиотека медицины США . Получено 7 февраля 2019 года .
- ^ Синдром Херлера в том, кто его назвал?
- ^ Hurler, G. (1919). «Через тип множественных адаптаций, в основном на скелетной системе» . Журнал педиатрии . 24 (5–6): 220–234. Doi : 10.1007/bf0222956 . S2CID 34471544 .