Белковая масс-спектрометрия

Белковая масс-спектрометрия относится к применению масс-спектрометрии для изучения белков . Масс-спектрометрия является важным методом точного определения массы и характеристики белков, и для его многочисленных применений было разработано множество методов и приборов. Его приложения включают идентификацию белков и их посттрансляционных модификаций , выяснение белковых комплексов, их субъединиц и функциональных взаимодействий, а также глобальное измерение белков в протеомике . Его также можно использовать для локализации белков в различных органеллах и определения взаимодействий между различными белками, а также с мембранными липидами. [1] [2]
Двумя основными методами, используемыми для ионизации белка в масс-спектрометрии, являются ионизация электрораспылением (ESI) и лазерная десорбция/ионизация с помощью матрицы (MALDI). Эти методы ионизации используются в сочетании с масс-анализаторами, такими как тандемная масс-спектрометрия . В общем, белки анализируются либо методом « сверху вниз », при котором белки анализируются неповрежденными, либо методом « снизу вверх », при котором белок сначала расщепляется на фрагменты. Иногда также может использоваться промежуточный подход «среднего вниз», при котором анализируются более крупные пептидные фрагменты.
История
[ редактировать ]Применение масс-спектрометрии для изучения белков стало популярным в 1980-х годах после разработки MALDI и ESI . Эти методы ионизации сыграли значительную роль в характеристике белков. (MALDI) Матричная ионизация лазерной десорбции была изобретена в конце 1980-х годов Францем Хилленкампом и Майклом Карасом . [3] Хилленкамп, Карас и их коллеги-исследователи смогли ионизировать аминокислоту аланин , смешав ее с аминокислотой триптофан и облучив импульсным лазером с длиной волны 266 нм. [3] Несмотря на свою важность, прорыв произошел только в 1987 году. В 1987 году Коичи Танака использовал «метод сверхтонкой металлической плюс жидкой матрицы» и ионизированные биомолекулы размером 34 472 Да протеинкарбоксипептидазы-А. [4]
В 1968 году Малкольм Доул сообщил о первом использовании ионизации электрораспылением с помощью масс-спектрометрии. Примерно в то же время, когда MALDI стал популяризирован, Джон Беннетт Фенн был упомянут как автор разработки ионизации электрораспылением. [5] [6] Коичи Танака получил Нобелевскую премию по химии 2002 года вместе с Джоном Фенном и Куртом Вютрихом «за разработку методов идентификации и структурного анализа биологических макромолекул». [7] Эти методы ионизации значительно облегчили исследование белков методом масс-спектрометрии. Следовательно, масс-спектрометрия белков теперь играет ведущую роль в характеристике белков.
Методы и подходы
[ редактировать ]Техники
[ редактировать ]Масс-спектрометрия белков требует, чтобы белки в растворе или твердом состоянии были переведены в ионизированную форму в газовой фазе перед их инъекцией и ускорением в электрическом или магнитном поле для анализа. Двумя основными методами ионизации белков являются ионизация электрораспылением (ESI) и лазерная десорбция/ионизация с помощью матрицы (MALDI). При электрораспылении ионы создаются из белков в растворе, что позволяет ионизировать хрупкие молекулы в целости и сохранности, иногда сохраняя нековалентные взаимодействия. В MALDI белки внедряются в матрицу, обычно в твердой форме, а ионы создаются импульсами лазерного света. Электроспрей производит больше многозарядных ионов, чем MALDI, что позволяет измерять белки с высокой массой и лучше фрагментировать их для идентификации, в то время как MALDI работает быстро и с меньшей вероятностью подвергается влиянию загрязняющих веществ, буферов и добавок. [8]
Массовый анализ цельного белка в основном проводится с использованием времяпролетной МС (TOF) или ионно-циклотронного резонанса с преобразованием Фурье (FT-ICR). Эти два типа приборов здесь предпочтительнее из-за их широкого диапазона масс, а в случае FT-ICR - высокой точности определения массы. Ионизация белка электрораспылением часто приводит к образованию нескольких заряженных частиц с 800 < m/z < 2000, и полученный спектр можно подвергнуть деконволюции для определения средней массы белка с точностью до 50 частей на миллион или лучше, используя TOF или инструменты с ионной ловушкой .
Массовый анализ протеолитических пептидов является популярным методом характеристики белков, поскольку для характеристики можно использовать более дешевые конструкции приборов. Кроме того, подготовка проб упрощается, когда цельные белки расщепляются на более мелкие пептидные фрагменты. Наиболее широко используемым инструментом для анализа массы пептидов являются инструменты MALDI-TOF, поскольку они позволяют получать отпечатки массы пептидов (PMF) с высокой скоростью (1 PMF можно проанализировать примерно за 10 секунд). Многоступенчатая квадрупольная времяпролетная система и квадрупольная ионная ловушка также находят применение в этом приложении.

Тандемная масс-спектрометрия (МС/МС) используется для измерения спектров фрагментации и идентификации белков с высокой скоростью и точностью. Диссоциация, вызванная столкновениями, используется в основных приложениях для создания набора фрагментов из определенного пептидного иона. Процесс фрагментации в первую очередь приводит к образованию продуктов расщепления, которые разрываются по пептидным связям. Из-за такой простоты фрагментации можно использовать наблюдаемые массы фрагментов для сопоставления с базой данных предсказанных масс для одной из многих заданных пептидных последовательностей. Тандемная МС ионов целого белка недавно была исследована с использованием диссоциации с электронозахватом и в принципе продемонстрировала обширную информацию о последовательностях, но это не является общепринятой практикой.
Подходы
[ редактировать ]В соответствии с характеристиками и массовым диапазоном доступных масс-спектрометров для характеристики белков используются два подхода. В первом случае интактные белки ионизируются любым из двух описанных выше методов, а затем вводятся в масс-анализатор. Этот подход называется стратегией анализа белков « сверху вниз », поскольку он предполагает начинать со всей массы, а затем разбирать ее на части. Однако нисходящий подход в основном ограничен исследованиями отдельных белков с низкой пропускной способностью из-за проблем, связанных с обработкой целых белков, их гетерогенностью и сложностью их анализа. [8]
Во втором подходе, называемом МС « снизу вверх », белки ферментативно расщепляются на более мелкие пептиды с использованием протеазы, такой как трипсин . Впоследствии эти пептиды вводят в масс-спектрометр и идентифицируют с помощью масс-дактилоскопии пептидов или тандемной масс-спектрометрии . Следовательно, этот подход использует идентификацию на уровне пептидов, чтобы сделать вывод о существовании белков, собранных вместе, с обнаружением повторов de novo . [9] Меньшие и более однородные фрагменты легче анализировать, чем интактные белки, и их также можно определить с высокой точностью, поэтому этот подход «снизу вверх» является предпочтительным методом исследований в протеомике. Еще один подход, который начинает приносить пользу, - это промежуточный подход «среднего вниз», при котором анализируются протеолитические пептиды, более крупные, чем типичные триптические пептиды. [8]
Фракционирование белков и пептидов
[ редактировать ]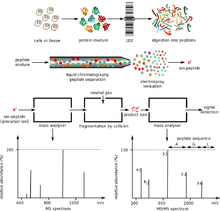
Представляющие интерес белки обычно являются частью сложной смеси множества белков и молекул, сосуществующих в биологической среде. Это создает две существенные проблемы. Во-первых, два метода ионизации, используемые для больших молекул, работают хорошо только тогда, когда смесь содержит примерно одинаковое количество материала, тогда как в биологических образцах разные белки обычно присутствуют в сильно различающихся количествах. Если такую смесь ионизировать с помощью электрораспыления или MALDI , более многочисленные виды имеют тенденцию «заглушать» или подавлять сигналы от менее распространенных. Во-вторых, масс-спектр сложной смеси очень трудно интерпретировать из-за подавляющего числа компонентов смеси. Это усугубляется тем, что ферментативное переваривание белка приводит к образованию большого количества пептидных продуктов.
В свете этих проблем для разделения белков широко используются методы одно- и двумерного гель-электрофореза и высокоэффективной жидкостной хроматографии. Первый метод фракционирует целые белки посредством двумерного гель-электрофореза . Первым измерением 2D-геля является изоэлектрическая фокусировка (ИЭФ). В этом измерении белок разделен по изоэлектрической точке (pI), а во втором измерении - электрофорез в ДСН-полиакриламидном геле (ДСН-ПААГ). Это измерение разделяет белок в соответствии с его молекулярной массой. [10] После завершения этого этапа происходит расщепление в геле. В некоторых ситуациях может потребоваться совмещение обоих этих методов. Гелевые пятна, обнаруженные на 2D-геле, обычно относятся к одному белку. Если требуется идентичность белка, обычно применяется метод расщепления в геле , при котором интересующее белковое пятно вырезается и переваривается протеолитически. Массы пептидов, полученные в результате расщепления, можно определить масс-спектрометрией с использованием дактилоскопии массы пептидов . Если эта информация не позволяет однозначно идентифицировать белок, его пептиды могут быть подвергнуты тандемной масс-спектрометрии для de novo секвенирования . Небольшие изменения массы и заряда можно обнаружить с помощью 2D-PAGE. Недостатками этого метода являются небольшой динамический диапазон по сравнению с другими методами, некоторые белки все еще трудно разделить из-за их кислотности, основности, гидрофобности и размера (слишком большого или слишком маленького). [11]
Второй метод, высокоэффективная жидкостная хроматография, используется для фракционирования пептидов после ферментативного расщепления. Характеризацию белковых смесей с помощью ВЭЖХ/МС также называют протеомикой дробовика и MuDPIT (технология многомерной идентификации белков). Пептидную смесь, полученную в результате расщепления смеси белков, фракционируют с помощью одной или двух стадий жидкостной хроматографии. Элюент со стадии хроматографии может быть либо непосредственно введен в масс-спектрометр посредством ионизации электрораспылением, либо нанесен на серию небольших пятен для последующего масс-анализа с использованием MALDI.
Приложения
[ редактировать ]Идентификация белка
[ редактировать ]Существует два основных способа использования MS для идентификации белков. При дактилоскопии пептидных масс массы протеолитических пептидов используются в качестве входных данных для поиска в базе данных прогнозируемых масс, которые могут возникнуть в результате переваривания списка известных белков. Если последовательность белка в списке ссылок приводит к значительному количеству предсказанных масс, соответствующих экспериментальным значениям, есть некоторые доказательства того, что этот белок присутствовал в исходном образце. Таким образом, этапы очистки ограничивают производительность метода массового дактилоскопирования пептидов. Альтернативно, пептиды можно фрагментировать с помощью МС/МС для более точной их идентификации. [9]
МС также является предпочтительным методом идентификации посттрансляционных модификаций белков по сравнению с другими подходами, такими как методы на основе антител. [1]
de novo (пептидов) Секвенирование
[ редактировать ]Секвенирование пептидов de novo для масс-спектрометрии обычно выполняется без предварительного знания аминокислотной последовательности. Это процесс выделения пептидных фрагментов белка из аминокислот . Секвенирование de novo оказалось успешным для подтверждения и расширения результатов поиска в базе данных.
Поскольку секвенирование de novo основано на массе, а некоторые аминокислоты имеют одинаковую массу (например, лейцин и изолейцин ), точное ручное секвенирование может быть затруднено. Следовательно, может оказаться необходимым использовать приложение для поиска гомологии последовательностей для совместной работы между поиском в базе данных и секвенированием de novo , чтобы устранить это неотъемлемое ограничение.
Преимущество поиска в базе данных заключается в быстром выявлении последовательностей при условии, что они уже задокументированы в базе данных. Другие неотъемлемые ограничения поиска в базе данных включают модификации/мутации последовательностей (некоторые поиски в базе данных не учитывают должным образом изменения «документированной» последовательности, поэтому могут быть упущены ценные сведения), неизвестность (если последовательность не задокументирована, она не будет найдена). ), ложные срабатывания, а также неполные и поврежденные данные. [12]
Аннотированную спектральную библиотеку пептидов также можно использовать в качестве эталона для идентификации белков/пептидов. Он предлагает уникальные преимущества: уменьшенное пространство поиска и повышенная специфичность. К ограничениям относятся: спектры, не включенные в библиотеку, не будут идентифицированы, спектры, собранные с разных типов масс-спектрометров, могут иметь весьма четкие особенности, а эталонные спектры в библиотеке могут содержать шумовые пики, что может привести к ложноположительной идентификации. [13] Был описан ряд различных алгоритмических подходов для идентификации пептидов и белков с помощью тандемной масс-спектрометрии (МС/МС), секвенирования пептидов de novo и поиска на основе меток последовательностей. [14]
Презентация антигена
[ редактировать ]Презентация антигена является первым шагом в обучении иммунной системы распознаванию новых патогенов. С этой целью антигенпрезентирующие клетки подвергают MHC иммунную систему фрагментам белка через молекулы . Однако не все фрагменты белка связываются с молекулами MHC конкретного человека. С помощью масс-спектрометрии можно определить истинный спектр молекул, представленных иммунной системе. [15]
Количественное определение белка
[ редактировать ]
Несколько методов позволяют количественно определять белки с помощью масс-спектрометрии. [16] а недавние достижения позволили количественно оценить тысячи белков в отдельных клетках. [17] [18] [19] Количественное определение белка с помощью масс-спектрометрии выигрывает от эффективного отбора проб (подсчета) многих ионов на белок по сравнению с другими методами. [20] [21] Количественные оценки могут выполняться методами без меток и мультиплексными методами, в которых в качестве меток используются изотопные массовые метки. Мультиплексные методы могут улучшить как количественную точность, так и пропускную способность. [22] [23] [24]
нерадиоактивные) более тяжелые изотопы углерода Обычно стабильные (например , ( 13 С) или азот ( 15 N) вводятся в один образец, а другой мечется соответствующими легкими изотопами (например, 12 С и 14 Н). [25] Перед анализом два образца смешивают. Пептиды, полученные из разных образцов, можно отличить по разнице в массе. Соотношение интенсивностей их пиков соответствует относительному соотношению пептидов (и белков). Первое поколение методов мечения изотопов включало SILAC (мечение стабильных изотопов аминокислотами в культуре клеток), катализируемое трипсином. 18 O-маркировка, ICAT (аффинная маркировка с изотопным кодированием) и iTRAQ (изобарические метки для относительного и абсолютного количественного анализа). [26] Более позднее поколение методов мультиплексирования включает тандемные массовые теги (TMT) для данных DDA и mTRAQ для мультиплексированного DIA (plexDIA). [27]
«Полуколичественную» масс-спектрометрию можно проводить без маркировки образцов. [28] Обычно это делается с помощью MALDI-анализа (в линейном режиме). Интенсивность пика или площадь пика отдельных молекул (обычно белков) здесь коррелирует с количеством белка в образце. Однако индивидуальный сигнал зависит от первичной структуры белка, сложности образца и настроек прибора. Другие типы количественной масс-спектрометрии «без меток» используют спектральные подсчеты (или подсчеты пептидов) переваренных белков как средство определения относительных количеств белка. [12]
Определение структуры белка
[ редактировать ]Характеристики, указывающие на трехмерную структуру белков, можно исследовать с помощью масс-спектрометрии различными способами. [29] Сравнение распределения зарядовых состояний может дать информацию о структуре белка. Большое разнообразие состояний с высоким зарядом указывает на беспорядок в белке, тогда как более компактные, свернутые белки приводят к состояниям с более низким зарядом. [30] Используя химическую сшивку для соединения частей белка, которые расположены близко в пространстве, но далеко друг от друга по последовательности, можно получить информацию об общей структуре. Следя за обменом амидных протонов на дейтерий из растворителя, можно проверить доступность растворителя для различных частей белка. [31] Водородно-дейтериевая обменная масс-спектрометрия используется для изучения белков и их конформаций уже более 20 лет. Этот тип структурного анализа белков может подойти для белков, которые являются сложными для других структурных методов. [32] Еще одним интересным направлением структурных исследований белков является ковалентное мечение, индуцированное лазером. В этом методе участки белка, подвергающиеся воздействию растворителя, модифицируются гидроксильными радикалами. Его сочетание с быстрым перемешиванием использовалось в исследованиях сворачивания белков. [33]
Протеогеномика
[ редактировать ]В том, что сейчас обычно называют протеогеномикой , пептиды, идентифицированные с помощью масс-спектрометрии, используются для улучшения аннотаций генов (например, стартовых сайтов генов) и аннотаций белков. Параллельный анализ генома и протеома облегчает обнаружение посттрансляционных модификаций и протеолитических событий. [34] особенно при сравнении нескольких видов. [35]
Ссылки
[ редактировать ]- ^ Jump up to: а б Юрген Кокс; Матиас Манн (июль 2011 г.). «Количественная протеомика высокого разрешения для системной биологии, управляемой данными» . Ежегодный обзор биохимии . 80 : 273–299. doi : 10.1146/annurev-biochem-061308-093216 . ПМИД 21548781 . – через Ежегодные обзоры (требуется подписка)
- ^ Нельсон П. Баррера; Кэрол В. Робинсон (июль 2011 г.). «Достижения в масс-спектрометрии мембранных белков: от отдельных белков к интактным комплексам». Ежегодный обзор биохимии . 80 : 247–71. doi : 10.1146/annurev-biochem-062309-093307 . hdl : 10533/135572 . ПМИД 21548785 . – через Ежегодные обзоры (требуется подписка)
- ^ Jump up to: а б Карась, М.; Бахманн, Д.; Хилленкамп, Ф. (1985). «Влияние длины волны в масс-спектрометрии органических молекул с ультрафиолетовой лазерной десорбцией при сильном излучении». Аналитическая химия . 57 (14): 2935–9. дои : 10.1021/ac00291a042 .
- ^ Карась, М.; Бахманн, Д.; Бахр, У.; Хилленкамп, Ф. (1987). «Матричная ультрафиолетовая лазерная десорбция нелетучих соединений». Международный журнал масс-спектрометрии и ионных процессов . 78 : 53–68. Бибкод : 1987IJMSI..78...53K . дои : 10.1016/0168-1176(87)87041-6 .
- ^ Доул М., Мак Л.Л., Хайнс Р.Л., Мобли Р.К., Фергюсон Л.Д., Элис М.Б. (1968). «Молекулярные пучки макроионов». Журнал химической физики . 49 (5): 2240–2249. Бибкод : 1968ЖЧФ..49.2240Д . дои : 10.1063/1.1670391 .
- ^ Бирендра Н. Праманик; А.К. Гангули; Майкл Л. Гросс (28 февраля 2002 г.). Прикладная масс-спектрометрия электрораспылением: серия практической спектроскопии . ЦРК Пресс . стр. 4–. ISBN 978-0-8247-4419-9 .
- ^ «Пресс-релиз: Нобелевская премия по химии 2002 г.» . Нобелевский фонд. 09.10.2002 . Проверено 2 апреля 2011 г.
- ^ Jump up to: а б с Чейт, Брайан Т. (2011). «Масс-спектрометрия в постгеномную эпоху». Анну Рев Биохим . 80 : 239–46. doi : 10.1146/annurev-biochem-110810-095744 . ПМИД 21675917 . – через Ежегодные обзоры (требуется подписка)
- ^ Jump up to: а б Траугер, А. Суния; В. Уэбб; Г. Сюздак (2002). «Анализ пептидов и белков методом масс-спектрометрии» . Спектроскопия . 16 (1): 15–28. дои : 10.1155/2002/320152 .
- ^ У. Уэллс, Г. Ван и С. Дж. Пэк (2006). «Сравнительное исследование трех протеомных количественных методов: DIGE, cICAT и iTRAQ, с использованием 2D-геля или LC-MALDI TOF/TOF». Журнал исследований протеома . 5 (3): 651–658. CiteSeerX 10.1.1.156.5802 . дои : 10.1021/pr050405o . ПМИД 16512681 .
- ^ Иссак, Дж. Халим и Т.Д. Винстра (2008). «Двумерный электрофорез в полиакриламидном геле (2D-PAGE): достижения и перспективы» . БиоТехники . 46 (5): 697–700. дои : 10.2144/000112823 . ПМИД 18474047 .
- ^ Jump up to: а б Брет, Купер, Дж. Фенг и В. Гарретт (2010). «Относительное количественное определение белка без меток: статистика ошибок спектрального подсчета на основе девяти повторных образцов MudPIT» . Спектроскопия . 21 (9): 1534–1546. дои : 10.1016/j.jasms.2010.05.001 . ПМИД 20541435 .
- ^ Огюстен, Скальберт, Л. Бреннан и О. Фин (2009). «Метаболомика на основе масс-спектрометрии: ограничения и рекомендации для будущего прогресса с особым акцентом на исследованиях в области питания» . Метаболомика . 5 (4): 435–458. дои : 10.1007/s11306-009-0168-0 . ПМЦ 2794347 . ПМИД 20046865 .
- ^ П. Эрнандес, М. Мюллер и Р.Д. Аппель (2006). «Автоматическая идентификация белков методом тандемной масс-спектрометрии: проблемы и стратегии». Обзоры масс-спектрометрии . 25 (2): 235–254. Бибкод : 2006MSRv...25..235H . дои : 10.1002/mas.20068 . ПМИД 16284939 .
- ^ Бузид Р., де Бейер М.Т., Луитен Р.Дж., Безстарости К., Кесслер А.Л., Бруно М.Дж., Пеппеленбош М.П., Деммерс Дж.А., Бушоу С.И. (май 2021 г.). «Эмпирическая оценка использования вычислительного связывания HLA в качестве раннего фильтра для рабочего процесса обнаружения эпитопов на основе масс-спектрометрии» . Раки . 13 (10): 2307. doi : 10.3390/cancers13102307 . ПМК 8150281 . ПМИД 34065814 .
- ^ Краватт, Бенджамин Ф.; Саймон, Габриэль М.; Йейтс III, Джон Р. (декабрь 2007 г.). «Биологическое воздействие протеомики, основанной на масс-спектрометрии» . Природа . 450 (7172): 991–1000. Бибкод : 2007Natur.450..991C . дои : 10.1038/nature06525 . ISSN 1476-4687 . ПМИД 18075578 . S2CID 205211923 .
- ^ Славов, Николай (01 февраля 2021 г.). «Анализ одноклеточных белков методом масс-спектрометрии» . Современное мнение в области химической биологии . 60 : 1–9. arXiv : 2004.02069 . дои : 10.1016/j.cbpa.2020.04.018 . ISSN 1367-5931 . ПМЦ 7767890 . ПМИД 32599342 .
- ^ Славов, Николай (31 января 2020 г.). «Распаковка протеома в одиночных клетках» . Наука . 367 (6477): 512–513. Бибкод : 2020Sci...367..512S . дои : 10.1126/science.aaz6695 . ISSN 0036-8075 . ПМК 7029782 . ПМИД 32001644 .
- ^ Хаффман, Р. Грей; Ледюк, Эндрю; Вихманн, Кристоф; Ди Джоя, Марко; Борриелло, Франческо; Шпехт, Харрисон; Деркс, Джейсон; Хан, Саад; Хури, Люк; Эммотт, Эдвард; Петельский, Александра А.; Перлман, Дэвид Х.; Кокс, Юрген; Занони, Иван; Славов, Николай (май 2023 г.). «Приоритет масс-спектрометрии увеличивает глубину, чувствительность и полноту данных протеомики отдельных клеток» . Природные методы . 20 (5): 714–722. дои : 10.1038/s41592-023-01830-1 . ISSN 1548-7091 . ПМЦ 10172113 . ПМИД 37012480 .
- ^ Маккосс, Майкл Дж.; Альфаро, Хавьер Антонио; Фавр, Даниэль А.; Ву, Кристина С.; Вануну, Мени; Славов, Николай (март 2023 г.). «Отбор проб протеома с помощью новых методов одиночной молекулы и масс-спектрометрии» . Природные методы . 20 (3): 339–346. дои : 10.1038/s41592-023-01802-5 . ISSN 1548-7105 . ПМЦ 10044470 . ПМИД 36899164 .
- ^ Славов, Николай (январь 2022 г.). «Подсчет белковых молекул для протеомики одиночных клеток» . Клетка . 185 (2): 232–234. дои : 10.1016/j.cell.2021.12.013 . ПМЦ 8855622 . ПМИД 35063071 .
- ^ Деркс, Джейсон; Славов, Николай (03 марта 2023 г.). «Стратегии увеличения глубины и производительности анализа белков с помощью plexDIA» . Журнал исследований протеома . 22 (3): 697–705. doi : 10.1021/acs.jproteome.2c00721 . ISSN 1535-3893 . ПМЦ 9992289 . ПМИД 36735898 .
- ^ Славов, Николай (январь 2022 г.). «Расширение масштабов протеомики отдельных клеток» . Молекулярная и клеточная протеомика . 21 (1): 100179. doi : 10.1016/j.mcpro.2021.100179 . ПМЦ 8683604 . ПМИД 34808355 .
- ^ «Система мультипликативного масштабирования протеомики одиночных клеток» . Природная биотехнология . 41 (1): 23–24. Январь 2023 г. doi : 10.1038/s41587-022-01411-1 . ISSN 1087-0156 . ПМИД 35851377 .
- ^ Снейдерс А.П., де Вос М.Г., Райт ПК (2005). «Новый подход к количественному определению и секвенированию пептидов на основе метаболического мечения 15N и 13C». J. Протеом Рез . 4 (2): 578–85. дои : 10.1021/pr0497733 . ПМИД 15822937 .
- ^ М. Мияги и ККС Рао (2007). «Протеолитический 18 Стратегии O-мечения для количественной протеомики». Обзоры масс-спектрометрии . 26 (1): 121–136. Bibcode : 2007MSRv...26..121M . doi : 10.1002/mas.20116 . PMID 17086517 .
- ^ Деркс, Джейсон; Ледюк, Эндрю; Вальманн, Георг; Хаффман, Р. Грей; Уиллетс, Мэтью; Хан, Саад; Шпехт, Харрисон; Ральсер, Маркус; Демичев Вадим; Славов, Николай (январь 2023 г.). «Увеличение производительности чувствительной протеомики с помощью plexDIA» . Природная биотехнология . 41 (1): 50–59. дои : 10.1038/s41587-022-01389-w . ISSN 1087-0156 . ПМЦ 9839897 . ПМИД 35835881 .
- ^ Хаккани А.С., Келли Дж.Ф., Станимирович Д.Б. (2008). «Количественное определение профиля белка методом масс-спектрометрии с использованием протеомики без меток». Протоколы геномики . Методы молекулярной биологии. Том. 439. стр. 241–56. дои : 10.1007/978-1-59745-188-8_17 . ISBN 978-1-58829-871-3 . ПМИД 18370108 .
- ^ З. Чжан; Д.Л. Смит (1994). «Изучение нековалентных структурных особенностей белков методом масс-спектрометрии». Обзоры масс-спектрометрии . 13 (5–6): 411–429. Бибкод : 1994MSRv...13..411S . дои : 10.1002/mas.1280130503 .
- ^ Суза, Анна К.; Ся, Цзыцзе; Тан, Генри YH; Тайнер, Джон А.; Уильямс, Эван Р. (01 февраля 2017 г.). «Зарядка белков в нативной масс-спектрометрии» . Журнал Американского общества масс-спектрометрии . 28 (2): 332–340. Бибкод : 2017JASMS..28..332S . дои : 10.1007/s13361-016-1517-7 . ISSN 1044-0305 . ПМЦ 5283922 . ПМИД 27734326 .
- ^ Т. Э. Уэльс и Дж. Р. Энген (2006). «Водородообменная масс-спектрометрия для анализа динамики белков». Обзоры масс-спектрометрии . 25 (1): 158–170. Бибкод : 2006MSRv...25..158W . дои : 10.1002/mas.20064 . ПМИД 16208684 .
- ^ Р.Э. Якоб и Дж.Р. Энген (2012). «Масс-спектрометрия водородного обмена: мы выбрались из зыбучих песков?» . Обзоры масс-спектрометрии . 23 (6): 1003–1010. Бибкод : 2012JASMS..23.1003I . дои : 10.1007/s13361-012-0377-z . ПМЦ 3389995 . ПМИД 22476891 .
- ^ BB Stocks и Л. Конерманн (2009). «Структурная характеристика короткоживущих промежуточных продуктов разворачивания белков с помощью лазерно-индуцированной окислительной маркировки и масс-спектрометрии». Анальный. хим. 81 (1): 20–27. дои : 10.1021/ac801888h . ПМИД 19055350 .
- ^ Гупта Н.; Таннер С.; Джейтли Н.; Адкинс Дж. Н.; Липтон М.; Эдвардс Р.; Ромин М.; Остерман А.; Бафна В.; Смит Р.Д.; и др. (2007). «Целопротеомный анализ посттрансляционных модификаций: применение масс-спектрометрии для протеогеномной аннотации» . Геном Рез . 17 (9): 1362–1377. дои : 10.1101/гр.6427907 . ПМК 1950905 . ПМИД 17690205 .
- ^ Гупта Н.; Бенхамида Дж.; Бхаргава В.; Гудман Д.; Каин Э.; Керман И.; Нгуен Н.; Олликайнен Н.; Родригес Х.; Ван Дж.; и др. (2008). «Сравнительная протеогеномика: сочетание масс-спектрометрии и сравнительной геномики для анализа множественных геномов» . Геном Рез . 18 (7): 1133–1142. дои : 10.1101/гр.074344.107 . ПМК 2493402 . ПМИД 18426904 .