Реакция Виттига
| Реакция Виттига | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Назван в честь | Георг Виттиг | ||||||||||
| Тип реакции | Реакция сцепления | ||||||||||
| Реакция | |||||||||||
| |||||||||||
| Условия | |||||||||||
| Типичные растворители | обычно ТГФ или диэтиловый эфир | ||||||||||
| Идентификаторы | |||||||||||
| марта Продвинутая органическая химия | 16–44 (6-е изд.) | ||||||||||
| Портал органической химии | реакция Виттига | ||||||||||
| RSC Идентификатор онтологии | RXNO: 0000015 | ||||||||||
| | |||||||||||
Реакция Виттига или олефинирование Виттига представляет собой химическую реакцию альдегида называемым или кетона трифенилфосфония, с илидом реагентом Виттига . Реакции Виттига чаще всего используются для превращения альдегидов и кетонов в алкены. [1] [2] [3] используют реакцию Виттига Чаще всего для введения метиленовой группы с помощью метилентрифенилфосфорана (Ph 3 P=CH 2 ). С помощью этого реагента даже стерически затрудненный кетон, такой как камфора, можно превратить в его метиленовое производное.

Механизм реакции [ править ]
Механистические исследования были сосредоточены на нестабилизированных илидах, поскольку промежуточные соединения можно отслеживать с помощью ЯМР-спектроскопии . Существование и взаимопревращение бетаина ( 3a и 3b ) являются предметом продолжающихся исследований. [4] Для реакций Виттига без лития исследования подтверждают согласованное образование оксафосфетана без вмешательства бетаина. В частности, илиды фосфония 1 реагируют с карбонильными соединениями 2 посредством [2+2] -циклоприсоединения , которое иногда описывают как имеющее топологию [ π 2 s + π 2 a ], с прямым образованием оксафосфетанов 4a и 4b . В условиях отсутствия лития стереохимия продукта 5 обусловлена кинетически контролируемым присоединением илида 1 к карбонилу 2 . Когда присутствует литий, может происходить уравновешивание промежуточных продуктов, возможно, через разновидности бетаина 3a и 3b . [5] [6] [7] Брюс Э. Марьянов и А.Б. Рейтц определили проблему уравновешивания промежуточных продуктов Виттига и назвали этот процесс «стереохимическим дрейфом». В течение многих лет предполагалось, что стереохимия реакции Виттига с точки зрения образования углерод-углеродных связей напрямую соответствует Z/E-стереохимии алкеновых продуктов. Однако некоторые реагенты не следуют этой простой схеме. Соли лития также могут оказывать глубокое влияние на стереохимический результат. [8]

Механизмы различаются для алифатических и ароматических альдегидов , а также для ароматических и алифатических илидов фосфония. Имеющиеся данные свидетельствуют о том, что реакция Виттига неразветвленных альдегидов в условиях, не содержащих солей лития, не уравновешивается и, следовательно, находится под кинетическим контролем реакции . [9] [10] Э. Ведейс выдвинул теорию, объясняющую стереоселективность стабилизированных и нестабилизированных реакций Виттига. [11]
Убедительные доказательства указывают на то, что в условиях без Li реакции Виттига с участием нестабилизированных (R 1 = алкил, H), полустабилизированных (R 1 = арил) и стабилизированных (R 1 = EWG) реагентов Виттига протекают через [2+2] /ретро-[2+2] механизм под кинетическим контролем, с оксафосфетаном в качестве единственного промежуточного продукта. [12]
Область применения и ограничения [ править ]
групповая Функциональная толерантность
Реактивы Виттига обычно допускают карбонильные соединения, содержащие несколько видов функциональных групп, таких как ОН , OR , нитроарены , эпоксиды , а иногда и сложные эфиры и амиды . [13] Даже кетоновые , альдегидные и нитрильные группы могут присутствовать, если они сопряжены с илидом — это упомянутые выше стабилизированные илиды . Бис-илиды (содержащие две связи P=C) также были получены и успешно использованы. [14] Может возникнуть проблема с стерически затрудненными кетонами, где реакция может быть медленной и давать плохие выходы, особенно со стабилизированными илидами, и в таких случаях реакция Хорнера-Уодсворта-Эммонса (HWE) предпочтительна (с использованием фосфонатных эфиров). Еще одним ограничением, о котором сообщается, является часто лабильная природа альдегидов , которые могут окисляться, полимеризоваться или разлагаться. В так называемом тандемном процессе окисления-Виттига альдегид образуется in situ путем окисления соответствующего спирта. [15]
Стереохимия [ править ]
Для реакции с альдегидами геометрию двойной связи легко предсказать на основании природы илида. С нестабилизированными илидами (R 3 = алкил), в результате чего образуется ( Z )-алкен с умеренной или высокой селективностью. Если реакцию проводят в диметилформамиде в присутствии йодида лития или йодида натрия , продукт почти исключительно представляет собой Z-изомер. [16] Со стабилизированными илидами (R 3 = сложный эфир или кетон), ( E )-алкен образуется с высокой селективностью. Селективность ( E )/( Z ) часто бывает плохой у полустабилизированных илидов (R 3 = арил). [17]
Для получения ( E )-алкена для нестабилизированных илидов можно использовать модификацию Шлоссера реакции Виттига. Альтернативно, олефинирование Юлия и его варианты также селективно обеспечивают ( E )-алкен. Обычно реакция Хорнера-Уодсворта-Эммонса дает ( E )-еноат (α,β-ненасыщенный сложный эфир), как и реакция Виттига. Для получения ( Z )-енолята можно использовать модификацию Стилла-Дженнари реакции Хорнера-Уодсворта-Эммонса.
Модификация Шлоссера [ править ]
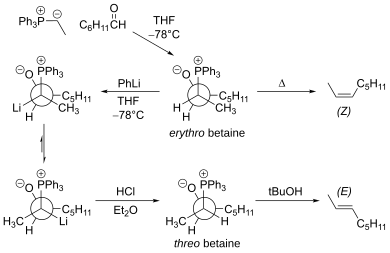
то, что реакция протекает в основном через промежуточный эритробетаин Основным ограничением традиционной реакции Виттига является , который приводит к Z-алкену. Эритробетаин можно превратить в треобетаин с помощью фениллития при низкой температуре. [18] Эта модификация дает E-алкен.
Аллиловые спирты можно получить реакцией бетаин-илида со вторым альдегидом. [19] Например:

Пример [ править ]
Примером его использования является синтез лейкотриена А. метилового эфира [20] [21] На первом этапе используется стабилизированный илид, где карбонильная группа сопряжена с илидом, предотвращая самоконденсацию, хотя неожиданно это дает в основном цис- продукт. Во второй реакции Виттига используется нестабилизированный реагент Виттига, и, как и ожидалось, в результате получается в основном цис- продукт.

История [ править ]
О реакции Виттига сообщили в 1954 году Георг Виттиг и его коллега Ульрих Шёллькопф . Частично за этот вклад Виттиг был удостоен Нобелевской премии по химии в 1979 году. [22] [23]
См. также [ править ]
- Реагент Кори – Чайковского
- Реакция Хорнера-Уодсворта-Эммонса
- Джулия олефинирование
- олефинирование по Петерсону
- реактив Теббе
- Фосфорорганическая химия
- Реакция омологации
- Олефинирование по Кауфману
- Метиленирование титана-цинка
Ссылки [ править ]
- ^ Maercker, A. Org. Реагировать. 1965 , 14 , 270–490.
- ^ В. Каррутерс, Некоторые современные методы органического синтеза , издательство Кембриджского университета, Кембридж, Великобритания, 1971, 81–90. ( ISBN 0-521-31117-9 )
- ^ Р.В. Хоффманн (2001). «Виттиг и его достижения: все еще актуальны после его 100-летия». Angewandte Chemie, международное издание . 40 (8): 1411–1416. doi : 10.1002/1521-3773(20010417)40:8<1411::AID-ANIE1411>3.0.CO;2-U . ПМИД 11317288 .
- ^ Э. Ведейс и К.Ф. Март (1990). «Механизм реакции Виттига: доказательства против промежуточных соединений бетаина». Дж. Ам. хим. Соц. 112 (10): 3905–3909. дои : 10.1021/ja00166a026 .
- ^ Брюс Э. Марьянофф , А. Б. Рейтц, М. С. Муттер, Р. Р. Иннерс и Х. Р. Алмонд-младший, «Подробные исследования скорости реакции Виттига нестабилизированных илидов фосфора с помощью 31 П, 1 Рука 13 С ЯМР-спектроскопия. Взгляд на кинетический и термодинамический контроль стереохимии», J. Am. Chem. Soc., 107 , 1068–1070 (1985).
- ^ Брюс Э. Марьянов, А.Б. Рейтц, Д.В. Граден и Х.Р. Алмонд-младший, «Исследование скорости ЯМР реакции Виттига 2,2-диметилпропаналя и трибутилбутилиденфосфорана», Tetrahedron Lett., 30 , 1361–1364 (1989). )
- ^ Брюс Э. Марьянофф, А. Б. Рейтц, М. С. Муттер, Р. Р. Иннерс, Х. Р. Алмонд-младший, Р. Р. Уиттл и Р. А. Олофсон, «Стереохимия и механизм реакции Виттига. Промежуточные продукты диастереомерной реакции и анализ хода реакции», J. Являюсь. хим. Соц., 108 , 7664–7678 (1986)
- ^ А.Б. Рейц, С.О. Норти, А.Д. Джордан-младший, М.С. Муттер и Брюс Э. Марьянофф, «Резкая концентрационная зависимость стереохимии в реакции Виттига. Исследование эффекта литий-соли», J. Org. хим., 51 , 3302–3308 (1986).
- ^ Э. Ведейс, К.Ф. Март и Р. Руджери (1988). «Эффекты заместителя и механизм Виттига: случай стереоспецифического разложения оксафосфетана». Дж. Ам. хим. Соц. 110 (12): 3940–48. дои : 10.1021/ja00220a036 .
- ^ Э. Ведейс и К.Ф. Март (1988). «Механизм реакции Виттига: роль заместителей у фосфора». Дж. Ам. хим. Соц . 110 (12): 3948–3958. дои : 10.1021/ja00220a037 .
- ^ Ведейс, Э.; Петерсон, М.Дж. Топ. Стереохим. 1994 , 21 , 1.
- ^ Бирн, Питер А.; Гилхиани, Деклан Г. (2013). «Современная интерпретация механизма реакции Виттига». Обзоры химического общества . 42 (16): 6670–96. дои : 10.1039/c3cs60105f . hdl : 10197/4939 . ISSN 0306-0012 . ПМИД 23673458 .
- ^ Смит (2020), Органическая химия марта , rxn. 16-44.
- ^ Б. Е. Марьянов и А. Б. Рейц (1989). «Реакция олефинирования Виттига и модификации с участием фосфорил-стабилизированных карбанионов. Стереохимия, механизм и некоторые аспекты синтеза». хим. Откр. 89 (4): 863–927. дои : 10.1021/cr00094a007 .
- ^ Ричард Дж. К. Тейлор, Леони Кэмпбелл и Грэм Д. Макаллистер (2008). «(±) транс-3,3'-(1,2-Циклопропандиил)бис-2-(E)-пропеновая кислота, диэтиловый эфир: процедура тандемного окисления (TOP) с использованием MnO 2 улавливания фосфорана, стабилизированного окислением » (PDF) . Органические синтезы . 85 : 15–26
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) . - ^ Л. Д. Бергельсон и М. М. Шемякин (1964). «Синтез встречающихся в природе ненасыщенных жирных кислот путем стерически контролируемого карбонильного олефинирования». Энджью. хим. 3 (4): 250–260. дои : 10.1002/anie.196402501 .
- ^ Робьетта, Рафаэль; Ричардсон, Джеффри; Аггарвал, Вариндер К.; Харви, Джереми Н. (1 февраля 2006 г.). «Реактивность и селективность реакции Виттига: вычислительное исследование». Журнал Американского химического общества . 128 (7): 2394–2409. дои : 10.1021/ja056650q . ISSN 0002-7863 . ПМИД 16478195 .
- ^ М. Шлоссер и К. Ф. Кристманн (1966). «Трансселективный синтез олефинов». Angewandte Chemie International Edition на английском языке . 5 (1): 126. дои : 10.1002/anie.196601261 .
- ^ Э. Дж. Кори и Х. Ямамото (1970). «Модификация реакции Виттига для обеспечения стереоспецифического синтеза некоторых тризамещенных олефинов. Стереоспецифический синтез α-санталола». Дж. Ам. хим. Соц. 92 (1): 226–228. дои : 10.1021/ja00704a052 .
- ^ И. Эрнест, А. Дж. Мейн и Р. Менасс (1982). «Синтез 7-цис-изомера природного лейкотриена d 4 ». Буквы тетраэдра . 23 (2): 167–170. дои : 10.1016/S0040-4039(00)86776-3 .
- ^ Э. Дж. Кори, Д. А. Кларк, Г. Гото, А. Марфат, К. Миосковски, Б. Самуэльссон и С. Хаммарстрём (1980). «Стереоспецифический тотальный синтез «медленно реагирующего вещества» анафилаксии, лейкотриена С-1». Дж. Ам. хим. Соц . 102 (4): 1436–1439. дои : 10.1021/ja00524a045 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Георг Виттиг, Ульрих Шёлькопф (1954). «О трифенилфосфинметилене как олефинобразующих реагентах I». Химические отчеты . 87 (9): 1318. doi : 10.1002/cber.19540870919 .
- ^ Георг Виттиг; Вернер Хааг (1955). «О трифенилфосфинметилене как олефинобразующих реагентах II». Химические отчеты . 88 (11): 1654–1666. дои : 10.1002/cber.19550881110 .
Внешние ссылки [ править ]
- Реакция Виттига в «Органическом синтезе» , Сб. Том. 10, с. 703 (2004); Том. 75, с. 153 (1998). ( Статья )
- Реакция Виттига в «Органическом синтезе» , Сб. Том. 5, с. 361 (1973); Том. 45, с. 33 (1965). ( Статья )
| Базы данных органов управления : Национальные |
|---|