Озонолиз
| Озонолиз | |
|---|---|
| Тип реакции | Органическая окислительно-восстановительная реакция |
| Идентификаторы | |
| Портал органической химии | механизм озонолиза-криджи |
В органической химии озонолиз — органическая реакция при которой ненасыщенные связи расщепляются озоном , ( О 3 ). Множественная связь углерод-углерод заменяется карбонильной ( C=O ) группы, такие как альдегиды, кетоны и карбоновые кислоты. Реакция преимущественно применяется к алкенам, но алкины и азосоединения также подвержены расщеплению. Исход реакции зависит от типа окисляемой кратной связи и условий обработки . [1]
Подробные процедуры описаны. [2] [3] [4]
Озонолиз алкенов
[ редактировать ]| Алкеновый озонолиз | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Тип реакции | Органическая окислительно-восстановительная реакция | ||||||||||
| Реакция | |||||||||||
| |||||||||||
| Идентификаторы | |||||||||||
| Портал органической химии | механизм озонолиза-криджи | ||||||||||
| RSC Идентификатор онтологии | RXNO: 0000344 | ||||||||||
Алкены могут окисляться озоном с образованием спиртов , альдегидов или кетонов или карбоновых кислот . В типичной процедуре озон барботируется через раствор алкена в метаноле при температуре -78 ° C (-108 ° F; 195 К), пока раствор не приобретет характерный синий цвет, обусловленный непрореагировавшим озоном. Однако промышленность рекомендует температуру около -20 ° C (-4 ° F; 253 К). [5] Такое изменение цвета указывает на полное израсходование алкена. Альтернативно, в качестве индикаторов этой конечной точки можно использовать различные другие реагенты путем обнаружения присутствия озона. Если озонолиз проводят путем введения через реакционную смесь потока обогащенного озоном кислорода, отходящий газ можно направить через раствор йодида калия . Когда раствор перестает поглощать озон, избыток озона окисляет йодид до йода , что легко заметить по его фиолетовому цвету. [6] Для более тщательного контроля самой реакции такой индикатор, как Судан Красный III в реакционную смесь можно добавить . Озон реагирует с этим показателем медленнее, чем с намеченной мишенью озонолиза. Озонолиз индикатора, вызывающий заметное изменение цвета, происходит только после того, как желаемая мишень будет израсходована. Если в субстрате имеются два алкена, которые реагируют с озоном с разной скоростью, можно выбрать индикатор, собственная скорость окисления которого является промежуточной между ними, и, следовательно, остановить реакцию, когда прореагировал только наиболее чувствительный алкен в субстрате. [7] В противном случае присутствие непрореагировавшего озона в растворе (его синий цвет) или в пузырьках (по данным обнаружения йодида) указывает только на то, что все алкены прореагировали.
После завершения добавления затем добавляют реагент для превращения промежуточного озонида в карбонильное производное. Восстановительные условия обработки используются гораздо чаще, чем окислительные.
Использование трифенилфосфина , тиомочевины , цинковой пыли или диметилсульфида приводит к образованию альдегидов или кетонов. При использовании боргидрида натрия образуются спирты. (Группа R также может представлять собой водороды)

Использование перекиси водорода может привести к образованию карбоновых кислот.

Амин- N -оксиды непосредственно производят альдегиды. [8] Другие функциональные группы , такие как бензиловые эфиры , также могут окисляться озоном. Было высказано предположение, что небольшие количества кислоты могут образовываться во время реакции в результате окисления растворителя, поэтому пиридин иногда используется для буферизации реакции. Дихлорметан часто используется в качестве сорастворителя в соотношении 1:1 для облегчения своевременного расщепления озонида. Азелаиновую кислоту и пеларгоновую кислоту получают озонолизом олеиновой кислоты в промышленных масштабах.
Примером может служить озонолиз эвгенола с превращением концевого алкена в альдегид: [9]

Контролируя условия реакции/обработки, из симметричных алкенов можно получить несимметричные продукты: [10]
- Использование ЦОН ; бикарбонат натрия (NaHCO 3 ); диметилсульфид (ДМС) дает альдегид и диметилацеталь .
- При использовании уксусного ангидрида (Ac 2 O) триэтиламин (Et 3 N) дает метиловый эфир и альдегид.
- Использование ЦОН; Ac 2 O, Et 3 N дает метиловый эфир и диметилацеталь.
Механизм реакции
[ редактировать ]
В общепринятом механизме, предложенном Рудольфом Криге в 1953 г., [11] [12] [13] алкен и озон образуют промежуточный молозонид в 1,3-диполярном циклоприсоединении . Затем молозонид превращается в соответствующий ему карбонильный оксид (также называемый промежуточным соединением Криджи или цвиттерионом Криджи ) и альдегид или кетон ( 3 ) в результате ретро-1,3-диполярного циклоприсоединения. Оксид и альдегид или кетон снова реагируют в виде 1,3-диполярного циклоприсоединения, образуя относительно стабильный промежуточный озонид , известный как триоксолан ( 4 ).

The reaction mechanism of ozonolysis.

Доказательства этого механизма можно найти в мечении изотопов . Когда 17 О-меченный бензальдегид реагирует с карбонильными оксидами, метка оказывается исключительно в эфирной связи озонида. [14] До сих пор ведутся споры о том, разрушается ли молозонид посредством согласованного или радикального процесса; это также может проявлять зависимость от субстрата.
История
[ редактировать ]Кристиан Фридрих Шенбейн , открывший озон в 1840 году, также совершил первый озонолиз: в 1845 году он сообщил, что этилен реагирует с озоном – после реакции не ощущался ни запах озона, ни запах этилена. [15] Озонолиз алкенов иногда называют «озонолизом Харриса», поскольку некоторые приписывают эту реакцию Карлу Дитриху Харрису . [16] До появления современных спектроскопических методов озонолиз был важным методом определения структуры органических молекул. Химики озонировали неизвестный алкен, чтобы получить более мелкие и легко идентифицируемые фрагменты.
Озонолиз алкинов
[ редактировать ]Озонолиз алкинов обычно дает кислый ангидрид или дикетоновый продукт. [17] не полная фрагментация, как у алкенов . Для этих реакций восстановитель не требуется. Механизм неизвестен. [18] Если реакцию проводят в присутствии воды, ангидрид гидролизуется с образованием двух карбоновых кислот .

Другие субстраты
[ редактировать ]исследуются редко Хотя азосоединения ( N=N ) подвержены озонолизу. Нитрозамины ( N−N=O ). [19]
Приложения
[ редактировать ]Основное применение озонолиза — преобразование ненасыщенных жирных кислот в производные с добавленной стоимостью. Озонолиз олеиновой кислоты является важным путем получения азелаиновой кислоты . Побочным продуктом является нонановая кислота : [20]
- CH 3 (CH 2 ) 7 CH=CH(CH 2 ) 7 CO 2 H} + 4 O 3 → HO 2 C(CH 2 ) 7 CO 2 H} + CH 3 (CH 2 ) 7 CO 2 H
Эруковая кислота является предшественником брассиловой кислоты , C13- дикарбоновой кислоты , которая используется для производства специальных полиамидов и полиэфиров . Превращение влечет за собой озонолиз, который избирательно расщепляет связь C=C в эруковой кислоте: [21]
- CH 3 (CH 2 ) 7 CH=CH(CH 2 ) 11 CO 2 H + O 3 + 0,5 O 2 → CH 3 (CH 2 ) 7 CO 2 H + HO 2 C(CH 2 ) 11 CO 2 H
Методом озонолиза получен ряд лекарств и их промежуточных продуктов. [22] Использование озона в фармацевтической промышленности трудно распознать из соображений конфиденциальности. [5]
Озонолиз как аналитический метод
[ редактировать ]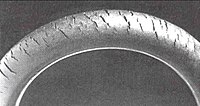
Озонолиз использовался для характеристики структуры некоторых полиолефинов . Ранние эксперименты показали, что повторяющейся единицей натурального каучука является изопрен .
возникновение
[ редактировать ]Озонолиз может стать серьезной проблемой, известной как озоновое растрескивание , когда следы газа в атмосфере разрушают эластомеры , такие как натуральный каучук , полибутадиен , бутадиен-стирол и нитриловый каучук . Озонолиз образует поверхностные кетоновые группы, которые могут вызвать дальнейшее постепенное разложение посредством реакций Норриша, если полимер подвергается воздействию света. Чтобы свести к минимуму эту проблему, многие продукты на основе полиолефина обрабатываются антиозонантами .
Озоновое растрескивание — это форма коррозионного растрескивания под напряжением , при которой активные химические вещества разъедают продукты чувствительного материала. Резиновое изделие должно находиться под напряжением , чтобы произошел рост трещин. Озоновое растрескивание когда-то часто наблюдалось на боковинах шин , где оно могло расшириться и вызвать опасный выброс , но сейчас это явление встречается редко из-за использования современных антиозонантов . Другие средства предотвращения включают замену чувствительных каучуков стойкими эластомерами, такими как полихлоропрен , EPDM или витон .
Безопасность
[ редактировать ]Использование озона в фармацевтической промышленности ограничено соображениями безопасности. [5]
См. также
[ редактировать ]- Деградация полимера
- Окисление Лемье-Джонсона - альтернативная система с использованием периодата и тетраоксида осмия.
- Trametes hirsuta — биотехнологическая альтернатива озонолизу.
Ссылки
[ редактировать ]- ^ Смит, Майкл Б.; Марч, Джерри (2007), Продвинутая органическая химия: реакции, механизмы и структура (6-е изд.), Нью-Йорк: Wiley-Interscience, стр. 1036, ISBN 978-0-471-72091-1
- ^ Бейли, PS; Эриксон, Р.Э. (1973). «Дифенальдегид» . Органические синтезы ; Сборник томов , т. 5, с. 489 .
- ^ Титце, LF; Братц, М. (1998). «Диалкилмезоксалаты путем озонолиза диалкилбензалмалонатов» . Органические синтезы ; Сборник томов , т. 9, с. 314 .
- ^ Харвуд, Лоуренс М.; Муди, Кристофер Дж. (1989). Экспериментальная органическая химия: принципы и практика (Иллюстрированное издание). Уайли-Блэквелл. стр. 55–57 . ISBN 978-0632020171 .
- ^ Перейти обратно: а б с Ван Орнум, Скотт Г.; Шампо, Робин М.; Париза, Ричард (2006). «Применение озонолиза в синтезе лекарств». Химические обзоры . 106 (7): 2990–3001. дои : 10.1021/cr040682z . ПМИД 16836306 .
- ^ Икан, Рафаэль (1991). Натуральные продукты: Лабораторное руководство (2-е изд.). Сан-Диего, Калифорния: Academic Press. п. 35. ISBN 0123705517 .
- ^ Вейсоглу, Тарик; Митчер, Лестер А.; Суэйзи, Джон К. (1980). «Удобный метод контроля селективного озонирования олефинов». Синтез . 1980 (10): 807–810. дои : 10.1055/s-1980-29214 .
- ^ Шварц, Крис; Райбл, Дж.; Мотт, К.; Дюссо, PH (2006). «Фрагментация карбонильных оксидов N -оксидами: улучшенный подход к озонолизу алкенов». Орг. Летт. 8 (15): 3199–3201. дои : 10.1021/ol061001k . ПМИД 16836365 .
- ^ Бранан, Брюс М.; Мясник, Джошуа Т.; Олсен, Лоуренс Р. (2007). «Использование озона в лаборатории органической химии: озонолиз эвгенола» . Дж. Хим. Образование. 84 (12): 1979. Бибкод : 2007JChEd..84.1979B . дои : 10.1021/ed084p1979 .
- ^ Клаус, Рональд Э.; Шрайбер, Стюарт Л. (1986). «Озонолитическое расщепление циклогексена до окончательно дифференцированных продуктов». Органические синтезы . 64 : 150. дои : 10.15227/orgsyn.064.0150 .
- ^ Криджи, Р. (1975). «Механизм озонолиза». Энджью. хим. Межд. Эд. англ. 14 (11): 745–752. дои : 10.1002/anie.197507451 .
- ^ «Механизм озонолиза» . Портал органической химии .
- ^ Ли, Цзе Джек (2006). «Механизм Криджи озонолиза». Назовите реакции . Спрингер. стр. 173–174. дои : 10.1007/3-540-30031-7_77 . ISBN 978-3-540-30030-4 .
- ^ Гелетнеки, К.; Бергер, С. (1998). «Механизм озонолиза, пересмотренный 17 O-ЯМР-спектроскопия». Eur. J. Org. Chem. 1998 (8): 1625–1627. doi : 10.1002/(SICI)1099-0690(199808)1998:8<1625::AID-EJOC1625>3.0.CO ;2-Л .
- ^ Кристиан Фридрих Шенбейн (1847 г.). «О поведении озона при образовании нефтеобразующего газа» . Отчет о переговорах Общества естественных исследований в Базеле (на немецком языке). 7 :7–9.
- ^ Мордехай Б. Рубин (2003). «История озона, часть III, К. Д. Харрис и введение озона в органическую химию» . Хелв. Хим. Акта . 86 (4): 930–940. дои : 10.1002/hlca.200390111 .
- ^ Бейли, PS (1982). «Глава 2». Озонирование в органической химии . Том. 2. Нью-Йорк, штат Нью-Йорк: Академик Пресс. ISBN 0-12-073102-9 .
- ^ Кремер, Д.; Креуэ, Р.; Англада, Дж. (2001). «Озонолиз ацетилена - квантово-химическое исследование». Дж. Ам. хим. Соц. 123 (25): 6127–6141. дои : 10.1021/ja010166f . ПМИД 11414847 .
- ^ Эндерс, Дитер; Киппхардт, Гельмут; Фей, Питер. «Асимметричные синтезы с использованием метода SAMP-/RAMP-гидрозона: ( S )-(+)-4-метил-3-гептанон» . Органические синтезы . 65 : 183. дои : 10.15227/orgsyn.065.0183 ; Сборник томов , т. 8, с. 403 .
- ^ Корнилс, Бой; Лаппе, Питер (2000). «Дикарбоновые кислоты алифатические». Энциклопедия промышленной химии Ульмана . дои : 10.1002/14356007.a08_523 . ISBN 978-3-527-30673-2 .
- ^ Аннекен, Дэвид Дж.; Оба, Сабина; Кристоф, Ральф; Фиг, Георг; Штайнбернер, Удо; Вестфехтель, Альфред (2006). «Жирные кислоты». Энциклопедия промышленной химии Ульмана . дои : 10.1002/14356007.a10_245.pub2 . ISBN 3527306730 .
- ^ Карон, Стефан; Даггер, Роберт В.; Руджери, Салли Гут; Рэган, Джон А.; Рипин, Дэвид Х. Браун (2006). «Крупномасштабное окисление в фармацевтической промышленности». Химические обзоры . 106 (7): 2943–2989. дои : 10.1021/cr040679f . ПМИД 16836305 .