Реакция Стилле
| Реакция Стилле | |
|---|---|
| Назван в честь | Джон Кеннет Стилл |
| Тип реакции | Реакция сцепления |
| Идентификаторы | |
| Портал органической химии | бесшумная муфта |
| RSC Идентификатор онтологии | RXNO: 0000035 |
Реакция Стилле — химическая реакция, широко используемая в органическом синтезе . Реакция включает в себя соединение двух органических групп, одна из которых представляет собой оловоорганическое соединение (также известное как оловоорганические соединения ). являются различные органические электрофилы Вторым партнером соединения . Реакция Стилле — одна из многих реакций сочетания, катализируемых палладием . [1] [2] [3]
- : Аллил, алкенил, арил, бензил, ацил.
- : галогениды (Cl, Br, I), псевдогалогениды (OTf, OPO(OR) 2 ), OAc
![{\displaystyle {\color {Blue}{\ce {R^{1}-Sn(Alkyl)3}}}+{\color {Red}{\ce {R^{2}-X}}}\ { \ce {->[{\color {Green}{\ce {Pd^{0}}}}{\text{ (каталитический)}}][{\text{набор лигандов}}]}}\ \overbrace { {\color {Blue}{\ce {R^{1}}}}\!-\!{\color {Red}{\ce {R^{2}}}}} ^{coupled\ product}+{ \color {Red}{\ce {X}}}\!-\!{\color {Blue}{\ce {Sn(Alkyl)3}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5baabb66db61c2d31fa2a5ca2b4e8156ee7c4133 "Общая схема реакции Стилле")


Р 1 группа, присоединенная к триалкилолово, обычно представляет собой sp 2 -гибридизированные, включающие винильную и арильную группы.
Эти станнаны также устойчивы как к воздуху, так и к влаге, и многие из этих реагентов либо коммерчески доступны, либо могут быть синтезированы на основании литературных данных. Однако эти оловянные реагенты, как правило, очень токсичны. X обычно представляет собой галогенид , такой как Cl , Br или I псевдогалогениды, такие как трифлаты , сульфонаты и фосфаты . , однако также можно использовать [4] [5] Опубликовано несколько обзоров. [6] [2] [7] [8] [9] [10] [11] [12] [13] [14] [15] [ чрезмерное цитирование ]
История
[ редактировать ]О первом примере катализируемого палладием сочетания арилгалогенидов с оловоорганическими реагентами сообщил Колин Иборн в 1976 году. [16] В результате этой реакции получали от 7% до 53% диарилового продукта. Этот процесс был расширен до сочетания ацилхлоридов с реагентами алкилолова в 1977 году Тошихико Мигитой, что дало от 53% до 87% кетонового продукта. [17]

В 1977 году Мигита опубликовал дальнейшую работу по сочетанию реагентов аллил -олова как с арил ( C ), так и с ацил ( D ) галогенидами. Большая способность аллильных групп мигрировать к палладиевому катализатору позволила проводить реакции при более низких температурах. Выходы арилгалогенидов составляли от 4% до 100%, ацилгалогенидов - от 27% до 86%. [18] [19] Отражая ранние вклады Мигиты и Косуги, реакцию Стилле иногда называют связью Мигиты-Косуги-Стилле .

Джон Кеннет Стилл впоследствии сообщил о сочетании различных реагентов алкилолова в 1978 году с многочисленными арил- и ацилгалогенидами в мягких условиях реакции с гораздо лучшими выходами (76–99%). [18] [20] В 1980-х годах Стилле продолжил свою работу по синтезу множества кетонов, используя этот широкий и мягкий процесс, и выяснил механизм этого преобразования. [21] [22]

К середине 1980-х годов было опубликовано более 65 статей по теме реакций сочетания с участием олова, в которых продолжалось изучение области субстратов этой реакции. Хотя первоначальные исследования в этой области были сосредоточены на связывании алкильных групп, большая часть будущих работ включала гораздо более полезное с синтетической точки зрения сочетание виниловых , алкенильных , арильных и аллилорганостаннанов с галогенидами. Благодаря устойчивости этих оловоорганических реагентов на воздухе и простоте их синтеза реакция Стилле стала распространенной в органическом синтезе. [8]
Механизм
[ редактировать ]Механизм реакции Стилле хорошо изучен. [11] [23] Каталитический цикл включает окислительное присоединение галогенида переметаллирование или псевдогалогенида ( 2 ) к палладиевому катализатору 1 ) , с образованием связанного 3 7 ( оловоорганическим реагентом 4 ) и восстановительное отщепление 5 ( продукта ( ) и регенерированного палладиевый катализатор ( 1 ). [24]

Однако подробный механизм сочетания Стилле чрезвычайно сложен и может происходить по многочисленным путям реакции. Как и другие реакции сочетания, катализируемые палладием , активным палладиевым катализатором считается 14-электронный комплекс Pd(0), который можно генерировать различными способами. Использование 18- или 16-электронного источника Pd(0) Pd(PPh 3 ) 4 , Pd(dba) 2 может подвергаться диссоциации лиганда с образованием активных частиц. Во-вторых, фосфины к безлигандному палладию(0) можно добавить . Наконец, как показано на рисунке, восстановление источника Pd(II) ( 8 ) (Pd(OAc) 2 , PdCl 2 (MeCN) 2 , PdCl 2 (PPh 3 ) 2 , BnPdCl(PPh 3 ) 2 и т. д.) путем добавления фосфиновых лигандов или оловоорганических реагентов. Также распространено использование [6]
Окислительное присоединение
[ редактировать ]Предложено окислительное присоединение к 14-электронному комплексу Pd(0). В результате этого процесса образуется 16-электронная разновидность Pd(II). Было высказано предположение, что анионные лиганды , такие как OAc , ускоряют этот этап за счет образования [Pd(OAc)(PR 3 ) n ] − , что делает виды палладия более нуклеофильными. [11] [25] В некоторых случаях, особенно когда сп 3 -гибридизированный органогалогенид , механизм типа S N 2 имеет тенденцию преобладать, однако это не так часто встречается в литературе. [11] [25] Однако, несмотря на то, что обычно образуется цис после согласованного окислительного присоединения -промежуточный продукт , этот продукт находится в быстром равновесии со своим транс -изомером. [26] [27]

Есть несколько причин, по которым изомеризация здесь предпочтительна . Во-первых, в этих процессах обычно используется объемный набор лигандов , например фосфины , и для них крайне неблагоприятно принимать цис -ориентацию относительно друг друга, что приводит к изомеризации в более выгодный транс-продукт. [26] [27] Альтернативное объяснение этого явления, получившего название антисимбиоза или трансфобии, заключается в упоминании СД. н модель. [24] [28] Согласно этой теории, палладий является гипервалентной разновидностью . Следовательно, Р 1 а транс-лиганд, будучи транс-по отношению друг к другу, будет конкурировать с одной орбиталью палладия за связь. Эта 4-электронная 3-центровая связь является самой слабой, когда присутствуют две сильные донорные группы, которые активно конкурируют за орбиталь палладия. По отношению к любому обычно используемому лиганду C-донор R 1 лиганд имеет гораздо более высокий транс-эффект . Это транс-влияние является мерой того, насколько конкурирующие транс-лиганды друг с другом будут конкурировать за орбиталь палладия. Обычный набор лигандов, фосфины и С-доноры (R 1 ) оба являются мягкими лигандами, а это означает, что они образуют прочные связи с палладием и активно конкурируют друг с другом за связь. [29] [30] Поскольку галогениды или псевдогалогениды значительно более электроотрицательны , их связь с палладием будет сильно поляризованной , при этом большая часть электронной плотности приходится на группу X, что делает их лигандами с низким транс-эффектом . Следовательно, это будет очень выгодно для R. 1 быть транс X, поскольку R 1 группа сможет образовать более прочную связь с палладием. [24] [28] [30]

Трансметаллизация
[ редактировать ]транс Считается, что трансметаллирование - промежуточного продукта на стадии окислительного присоединения происходит по различным механизмам в зависимости от субстратов и условий. Наиболее распространенный тип трансметаллирования для связи Стилле включает ассоциативный механизм . Этот путь подразумевает, что станнан , обычно атом олова , связанный с аллильной, алкенильной или арильной группой, может координироваться с палладием через одну из этих двойных связей. В результате образуется мимолетная пятивалентная 18-электронная разновидность , которая затем может подвергнуться отсоединению лиганда, чтобы снова сформировать плоский квадратный комплекс. Несмотря на то, что станнан органостанан координируется с палладием через R 2 группа, Р 2 формально должен быть переведен в палладий (R 2 -Sn связь должна быть разорвана), а группа X должна уйти вместе с оловом, завершив трансметаллирование. Считается, что это происходит посредством двух механизмов. [31]
Во-первых, когда станнан первоначально присоединяется к транс-металлическому комплексу, группа X может координироваться с оловом , в дополнение к палладию, создавая циклическое переходное состояние . Распад этого аддукта приводит к потере R 3 Sn-X и комплекса трехвалентного палладия с R 1 и Р 2 присутствует в цис- отношениях. Другой часто встречающийся механизм включает в себя то же первоначальное добавление органостаннана к транспалладиевому комплексу, как показано выше; однако в этом случае группа X не координируется с оловом, образуя открытое переходное состояние . После того, как α-углерод по отношению к олову атакует палладий, комплекс олова останется с чистым положительным зарядом. Обратите внимание, что на схеме ниже двойная связь, координирующая олово, обозначает R. 2 , то есть любая алкенильная , аллильная или арильная группа. Более того, группа X может диссоциировать в любой момент механизма и связываться с Sn. + комплекс в конце. Расчеты теории функционала плотности предсказывают, что открытый механизм будет преобладать, если два лиганда останутся прикрепленными к палладию и листьям группы X, тогда как циклический механизм более вероятен, если лиганд диссоциирует до трансметаллирования . Следовательно, хорошие уходящие группы, такие как трифлаты, в полярных растворителях отдают предпочтение циклическому переходному состоянию, тогда как объемистые фосфиновые лиганды предпочитают открытое переходное состояние. [31]

Менее распространенный путь трансметаллирования - это диссоциативный механизм или механизм с участием растворителя. Здесь лиганд четырехвалентного палладия диссоциирует, и к палладию может быть добавлен координирующий растворитель. Когда растворитель отсоединяется с образованием трехвалентного промежуточного соединения с 14 электронами, станнан может присоединиться к палладию , подвергаясь процессу открытого или циклического типа, как указано выше. [31]
Шаг восстановительного исключения
[ редактировать ]Для того, чтобы Р 1 -Р 2 для редукционного устранения эти группы должны занимать взаимно цис- координационные места. Поэтому любые транс -аддукты должны изомеризоваться с цис- промежуточным соединением, иначе связывание будет нарушено. Существует множество механизмов редукционной элиминации, и они обычно считаются согласованными. [11] [32] [33]
Во-первых, 16-электронный четырехвалентный промежуточный продукт со стадии трансметаллирования может подвергаться самостоятельному восстановительному элиминированию из плоско-квадратного комплекса. Эта реакция протекает в два этапа: сначала восстановительное отщепление, за которым следует координация вновь образовавшейся сигма-связи между R 1 и Р 2 к металлу, с окончательной диссоциацией с образованием связанного продукта. [11] [32] [33]

Однако предыдущий процесс иногда протекает медленно и может быть значительно ускорен за счет диссоциации лиганда с образованием 14-электронного Т-образного промежуточного продукта . Затем этот промежуточный продукт может перегруппироваться с образованием Y-образного аддукта, который может подвергаться более быстрому восстановительному элиминированию. [11] [32] [33]

Наконец, дополнительный лиганд может ассоциироваться с палладием с образованием 18-электронной тригональной бипирамидальной структуры с R 1 и Р 2 цис друг к другу в экваториальных положениях. Геометрия этого промежуточного продукта делает его похожим на Y-образный, описанный выше. [11] [32] [33]

Присутствие объемистых лигандов также может увеличить скорость элиминации. Лиганды, такие как фосфины , с большими углами прикуса вызывают стерическое отталкивание между L и R. 1 и Р 2 , в результате чего угол между группами L и R увеличивается, а угол между группами R 1 и Р 2 следовательно, уменьшиться, что позволит ускорить восстановительную элиминацию . [11] [24]

Кинетика
[ редактировать ]Скорость трансметаллирования оловоорганических соединений на палладиевых катализаторах показана ниже. СП 2 -гибридизированные углеродные группы, присоединенные к олову, являются наиболее часто используемыми партнерами сочетания, а sp 3 -гибридизированные углероды требуют более жестких условий, а концевые алкины могут соединяться через связь CH посредством реакции Соногаширы .

В качестве оловоорганического соединения обычно используют триметилстаннил или трибутилстаннил. Хотя триметилстанниловые соединения проявляют более высокую реакционную способность по сравнению с трибутилстанниловыми соединениями и имеют гораздо более простое 1 В спектрах H-ЯМР токсичность первого значительно выше. [34]
Оптимизация того, какие лиганды лучше всего подходят для проведения реакции с высоким выходом и скоростью оборота, может быть затруднена. Это связано с тем, что для окислительного присоединения требуется металл, богатый электронами, поэтому предпочтение отдается лигандам, отдающим электроны. Однако электронодефицитный металл более благоприятен для стадий трансметаллирования и восстановительного элиминирования , что делает электроноакцепторные лиганды лучшими здесь. Следовательно, оптимальный набор лигандов во многом зависит от индивидуальных субстратов и используемых условий. Это может изменить стадию определения скорости, а также механизм стадии трансметаллирования . [35]
Обычно используют лиганды промежуточной доности, такие как фосфины. Увеличение скорости можно увидеть при использовании лигандов с умеренной электронами, таких как три-2-фурилфосфин или трифениларсенин. Аналогичным образом, лиганды с большим количеством доноров могут замедлять или ингибировать реакции сочетания. [35] [36]
Эти наблюдения подразумевают, что обычно стадией, определяющей скорость реакции Стилле, является трансметаллирование . [36]
Добавки
[ редактировать ]Наиболее распространенной добавкой к реакции Стилле является стехиометрическая или сокаталитическая медь(I) , в частности йодид меди , который может повысить скорость реакции более чем на 10 раз. 3 складывать. Было высказано предположение, что в полярных растворителях медь трансметаллируется с органостаннаном . Полученный органокупратный реагент затем может трансметаллироваться с палладиевым катализатором. Кроме того, в эфирных растворителях медь также может способствовать удалению фосфинового лиганда , активируя Pd-центр. [9] [37] [38] [39] [40]
хлорид лития Было обнаружено, что трансметаллирования , является мощным ускорителем скорости в случаях, когда группа X диссоциирует от палладия (т.е. по открытому механизму). Считается, что хлорид-ион либо замещает группу X в палладии, делая катализатор более активным в отношении либо координируется с аддуктом Pd(0) для ускорения окислительного присоединения . Кроме того, соль LiCl усиливает полярность растворителя, облегчая этого обычно анионного лиганда (–Cl , –Br , –OTf и выход т. д.). Эта добавка необходима при такого растворителя, как ТГФ использовании ; однако использование более полярного растворителя, такого как NMP , может заменить необходимость в этой солевой добавке. Однако когда стадия трансметаллирования протекает по циклическому механизму, добавление хлорида лития может фактически снизить скорость. Как и в циклическом механизме, вместо анионной группы X должен диссоциировать нейтральный лиганд, например фосфин. [10] [41]
также влияют источники фторид-ионов , такие как фторид цезия Наконец, на каталитический цикл . Во-первых, фторид может увеличивать скорость реакций органотрифлатов , возможно, за счет того же эффекта, что и хлорид лития . Кроме того, ионы фтора могут действовать как поглотители побочных олова продуктов , что облегчает их удаление фильтрацией . [39]
Конкурирующие побочные реакции
[ редактировать ]Наиболее распространенной побочной реакционной способностью, связанной с реакцией Стилле, является гомосочетание станнановых реагентов с образованием R. 2 -Р 2 димер . Считается, что это происходит посредством двух возможных механизмов. Во-первых, реакция двух эквивалентов станнана приведет к получению гомосопряженного продукта с предкатализатором Pd (II) после восстановительного элиминирования . Во-вторых, катализатор Pd(0) может подвергнуться радикальному процессу с образованием димера. Используемый станнановый реагент традиционно является четырехвалентным по олову и обычно состоит из sp. 2 -гибридизированная группа, подлежащая переносу, и три «непереносимые» алкильные группы. Как видно выше, алкильные группы обычно мигрируют на палладиевый катализатор медленнее всего. [10]

Также было обнаружено, что при температуре всего 50 ° C арильные группы как на палладии , так и на координированном фосфине могут обмениваться. Хотя обычно они не обнаруживаются, во многих случаях они могут быть потенциальным второстепенным продуктом. [10]
Наконец, довольно редкая и экзотическая побочная реакция известна как кинозамещение . Здесь, после первоначального окислительного присоединения арилгалогенида , эта разновидность Pd-Ar может внедряться по двойной связи винилового олова. После удаления β-гидрида , миграционной вставки и протодестаннирования можно синтезировать 1,2-дизамещенный олефин. [10]

Могут возникнуть многочисленные другие побочные реакции, в том числе E/Z-изомеризация , которая потенциально может стать проблемой при использовании алкенилстаннана. Механизм этой трансформации в настоящее время неизвестен. Обычно станнаны-органические соединения довольно устойчивы к гидролизу , однако при использовании арилстаннанов с очень высоким содержанием электронов это может стать существенной побочной реакцией. [10]
Объем
[ редактировать ]электрофил
[ редактировать ]Винилгалогениды являются обычными партнерами реакции Стилле, и реакции этого типа встречаются в многочисленных процессах природных продуктов полного синтеза . Обычно используются винилиодиды и бромиды. Винилхлориды недостаточно активны в отношении окислительного присоединения к Pd(0). Обычно предпочтение отдается йодидам : они обычно реагируют быстрее и в более мягких условиях, чем бромиды . Это различие продемонстрировано ниже на примере селективного сочетания винилиодида в присутствии винилбромида. [10]
Обычно стереохимия алкена . сохраняется на протяжении всей реакции, за исключением суровых условий реакции Могут быть использованы различные алкены, в том числе α- и β-галоген-α, β-ненасыщенные кетоны , сложные эфиры и сульфоксиды (для работы которых обычно требуется добавка меди (I)) и многое другое (см. пример ниже). . [42] Также иногда используются виниловые трифлаты. Некоторые реакции требуют добавления LiCl , а другие замедляются, что означает наличие двух механизмов. [10]

Другим классом распространенных электрофилов являются арил- и гетероциклические галогениды. Что касается виниловых подложек, то бромиды и йодиды более распространены, несмотря на их большую стоимость. Можно выбрать множество арильных групп, включая кольца, замещенные электронодонорными заместителями, биарильные кольца и т. д. галогензамещенные гетероциклы В качестве партнеров сочетания также использовались , включая пиридины , фураны , тиофены , тиазолы , индолы , имидазолы , пурины , урацилы , цитозины , пиримидины и другие (см. таблицу гетероциклов ниже; галогены могут быть заменены на различные позиций по каждому). [10]

приведен пример использования реакции Стилле для усложнения гетероциклов нуклеозидов Ниже , таких как пурины . [43]

Арилтрифлаты сульфонаты и . также сочетаются с широким спектром станнановых реагентов Трифлаты имеют тенденцию реагировать аналогично бромидам в реакции Стилле. [10]
Ацилхлориды также используются в качестве партнеров сочетания и могут использоваться с широким спектром станнановых соединений, даже с реагентами на основе алкилолова, для получения кетонов (см. пример ниже). [44] ацилхлорида Однако иногда бывает трудно ввести функциональные группы в большие молекулы с чувствительными функциональными группами. Альтернативой этому процессу является реакция карбонилирующего кросс-сочетания Стилле, которая вводит карбонильную группу посредством внедрения монооксида углерода . [10]

аллильные , бензильные и пропаргиловые Также могут быть связаны галогениды. Хотя обычно используются, аллильные галогениды действуют через η 3 переходное состояние, позволяющее связываться с органостаннаном либо в положении α, либо в положении γ, происходящем преимущественно у наименее замещенного углерода (см. пример ниже). [45] Алкенилэпоксиды (соседние эпоксиды и алкены ) также могут подвергаться такому же связыванию через η 3 переходное состояние , например, открытие эпоксида в спирт . аллиловые и бензиловые ацетаты , ацетаты пропаргиловой кислоты не реагируют с органостаннанами. Хотя обычно используются [10]

Станнан
[ редактировать ]Станнановые органические реагенты широко распространены. Некоторые из них имеются в продаже. [46] Станнановые реагенты можно синтезировать реакцией реактива Гриньяра или литийорганического реагента с хлоридами триалкилолова. Например, винилтрибутилолово получают реакцией винилмагнийбромида с хлоридом трибутилолова . [47] Гидростаннилирование алкинов алкенов или дает множество производных. Оловоорганические реагенты устойчивы к воздуху и влаге. Некоторые реакции могут протекать даже в воде. [48] Их можно очистить хроматографией . Они толерантны к большинству функциональных групп. Некоторые оловоорганические соединения сильно токсичны , особенно производные триметилстаннила. [10]
Широко распространено использование винилстаннановых или алкенилстаннановых реагентов. [10] Что касается ограничений, то как очень объемистые станнановые реагенты, так и станнаны с замещением α-углерода имеют тенденцию реагировать вяло или требуют оптимизации. Например, в приведенном ниже случае α-замещенный винилстаннан реагирует только с концевым йодидом из-за стерических затруднений . [49]
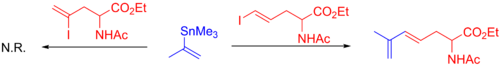
Арилстаннановые реагенты также распространены, и как электронодонорные , так и электроноакцепторные группы фактически увеличивают скорость трансметаллирования. Это снова означает, что могут иметь место два механизма трансметаллирования . Единственным ограничением для этих реагентов являются заместители в орто-положении, настолько маленькие, что метильные группы могут снизить скорость реакции. самые разнообразные гетероциклы В качестве партнеров сочетания также можно использовать (см. раздел «Электрофилы») (см. пример с тиазольным кольцом ниже). [10] [50]


Алкинилстаннаны, наиболее реакционноспособные из станнанов, также использовались в муфтах Стилле. Обычно в них нет необходимости, поскольку концевые алкины могут напрямую связываться с палладиевыми катализаторами через связь CH посредством сочетания Соногашира . Сообщается, что аллилстаннаны эффективны, однако возникают трудности, как и в случае с аллилгалогенидами, связанные с трудностью контроля региоселективности присоединения α и γ. Реагенты дистаннан и ацилстаннан также использовались в соединениях Стилле. [10]
Приложения
[ редактировать ]Реакция Стилле использовалась в синтезе различных полимеров. [51] [52] [53] Однако наиболее широкое применение реакции Стилле — это ее применение в органических синтезах , и конкретно — в синтезе природных продуктов .
Полный синтез натуральных продуктов
[ редактировать ]Ларри Овермана 19-ступенчатый энантиоселективный полный синтез квадригемина С включает двойную реакцию перекрестного метатезиса Стилле . [6] [54] Комплекс станнанорганического соединения связан с двумя арилиодидными группами. После двойной циклизации Хека получают продукт.

(+)-микотриенола Панека 32-ступенчатом энантиоселективном полном синтезе ансамицинового антибиотика В используется поздней стадии тандемного соединения макроциклов типа Стилле. Здесь станнан имеет две концевые группы трибутилолова, атакованные алкеном. Этот станнан «сшивает» два конца линейного исходного материала в макроцикл, добавляя при этом недостающие два метиленовых звена. После окисления ароматического ядра нитратом церия-аммония (CAN) и снятия защиты получается плавиковой кислотой натуральный продукт с выходом 54% на 3 стадиях. [6] [55]

В 21-этапном энантиоселективном полном синтезе противоопухолевого алкалоида манзамина Ирцинал А Стивена Ф. Мартина и его коллег используется тандемная однореакторная реакция Стилле/Дильса-Альдера. Алкеновая группа добавляется к винилбромиду с последующим in situ Дильса-Альдера циклоприсоединением между добавленным алкеном и алкеном в пирролидиновом кольце. [6] [56]

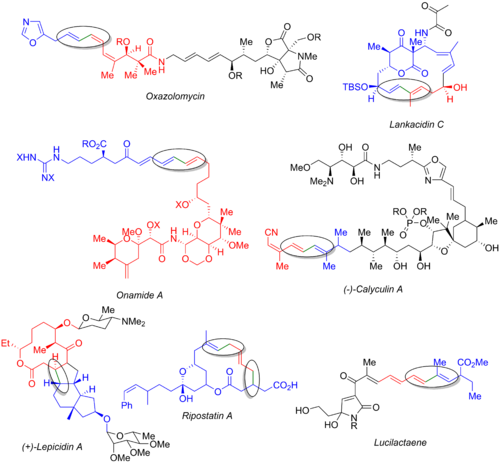
Во многих других методах полного синтеза используется реакция Стилле, включая реакции оксазоломицина, [57] ланкацидин С, [58] онамид А, [59] каликулин А, [60] лепицидин А, [61] рипостатин А, [62] и люцилактаен. [6] [63] На изображении ниже показан конечный природный продукт : органогалогенид (синий), станнан (красный) и образующаяся связь (зеленый и обведенный кружком). Из этих примеров видно, что реакцию Стилле можно использовать как на ранних стадиях синтеза (оксазоломицин и каликулин А), так и в конце конвергентного пути (онамид А, ланкацидин С, рипостатин А) или в средний (лепицидин А и люцилактаен). Синтез рипостатина А включает два одновременных связывания Стилле, за которыми следует метатезис с замыканием кольца . Синтез люцилактаена включает среднюю субъединицу, имеющую боран с одной стороны и станнан с другой, что позволяет провести реакцию Стилле с последующим сочетанием Сузуки.
Вариации
[ редактировать ]Помимо проведения реакции в различных органических растворителях, были разработаны условия, которые позволяют проводить широкий диапазон реакций Стилле в водном растворителе. [14]
Было показано , что в присутствии солей Cu(I) палладий на угле является эффективным катализатором. [64] [65]
В области зеленой химии сообщается, что реакция Стилле протекает в легкоплавкой и высокополярной смеси сахара, такого как маннит , мочевины, такой как диметилмочевина, и соли, такой как хлорид аммония. [66] . [67] Каталитическая система представляет собой трис(дибензилиденацетон)дипалладий(0) с трифениларсином :

Стилле-карбонильное кросс-сочетание
[ редактировать ]Распространенным изменением реакции Стилле является включение карбонильной группы между R 1 и Р 2 , служащий эффективным методом образования кетонов . Этот процесс чрезвычайно похож на первоначальные исследования Мигиты и Стилле (см. «Историю») по соединению станнана с ацилхлоридами . Однако эти фрагменты не всегда легко доступны, и их может быть трудно сформировать, особенно в присутствии чувствительных функциональных групп . Кроме того, контроль их высокой реакционной способности может оказаться сложной задачей. атмосфера монооксида углерода В реакции Стилле-карбонилативного кросс-сочетания используются те же условия, что и в реакции Стилле, за исключением того, что используется (CO). CO может координироваться с палладиевым катализатором ( 9 ) после первоначального окислительного добавления с последующим внедрением CO в Pd-R. 1 связь ( 10 ), что приводит к последующему восстановительному отщеплению до кетона ( 12 ). Стадия трансметаллирования обычно является стадией, определяющей скорость . [6]

Ларри Оверман коллеги используют карбонильную перекрестную связь Стилле в своем 20-этапном энантиоселективном полном синтезе стрихнина и его . Добавленный карбонил позже превращается в терминальный алкен посредством реакции Виттига , что позволяет сформировать ключевой третичный азот и пентациклическое ядро посредством аза- Коупа - реакции Манниха . [6] [68]
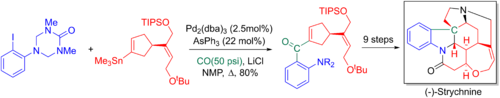
Джорджио Ортар и др. исследовал, как можно использовать карбонильную кросс-сочетание Стилле для синтеза бензофеноновых фосфоров. 4-бензоил-L-фенилаланина Они были встроены в пептиды и использованы из-за их фотоаффинных свойств мечения для изучения различных пептид-белковых взаимодействий. [6] [69]

16-ступенчатый полный рацемический синтез ятрафона Луи Хегедуса включал карбонилирующее кросс-сочетание Стилле в качестве последнего этапа формирования 11-членного макроцикла . Вместо галогенида здесь в качестве партнера сочетания используется винилтрифлат. [6] [70]

Связь Стилле – Келли
[ редактировать ]Используя плодотворную публикацию Иборна в 1976 году, в которой арилстаннаны образуются из арилгалогенидов и дистаннанов, Т. Росс Келли применил этот процесс к внутримолекулярному сочетанию арилгалогенидов. Это тандемное сочетание станнилирование/арилгалогенид использовалось для синтеза различных дигидрофенантренов. Большинство образующихся внутренних колец ограничено 5 или 6 членами, однако сообщалось о некоторых случаях макроциклизации. В отличие от обычного сочетания Стилле, хлор не действует как галоген, возможно, из-за его более низкой реакционной способности в последовательности галогенов (его более короткая длина связи и более сильная энергия диссоциации связи затрудняют разрыв посредством окислительного присоединения ). Начиная с середины схемы ниже и двигаясь по часовой стрелке, палладиевый катализатор ( 1 ) окислительно присоединяется к наиболее реакционноспособной связи CX ( 13 ) с образованием 14 , за которым следует трансметаллирование дистаннаном ( 15 ) с получением 16 и восстановительным элиминированием с образованием соединения 14. арилстаннан ( 18 ). Регенерированный палладиевый катализатор ( 1 ) может подвергаться окислительному добавлению. ко второй связи CX 18 с образованием 19 с последующим внутримолекулярным трансметаллированием с получением 20 с последующим восстановительным элиминированием с получением связанного продукта ( 22 ). [6]

Цзе Джек Ли и др. использовали реакцию Стилле-Келли в синтезе различных кольцевых систем бензо[4,5]фуропиридинов. Они используют трехэтапный процесс, включающий аминирование Бухвальда-Хартвига , еще одну реакцию сочетания, катализируемую палладием , за которой следует внутримолекулярное сочетание Стилле-Келли. Обратите внимание, что арил-йодидная связь будет окислительно присоединяться к палладию быстрее, чем любая из арил-бромидных связей. [6] [71]
![Синтез бензо[4,5]фуропиридинов](http://upload.wikimedia.org/wikipedia/commons/thumb/2/25/Benzofuropyridines.png/500px-Benzofuropyridines.png)
См. также
[ редактировать ]- Оловоорганическая химия
- Органостаннановая добавка
- Реакции сочетания, катализируемые палладием
- Реакция Сузуки
- Муфта Негиши
- Черт возьми реакция
- Хияма муфта
Ссылки
[ редактировать ]- ^ Хартвиг, Дж. Ф. Химия органопереходных металлов, от связывания к катализу ; Университетские научные книги: Нью-Йорк, 2010. ISBN 189138953X
- ^ Перейти обратно: а б Стилле, Дж. К. Энджью. хим. Межд. Эд. англ. 1986 , 25 , 508–524. ( Обзор )
- ^ Фарина, В.; Кришнамурти, В.; Скотт, WJ Org. Реагировать. 1998 , 50 , 1–652. ( Обзор )
- ^ Скотт, WJ; Крисп, GT; Стилле, Дж. К. «Органические синтезы » , Coll. Том. 8, с. 97 (1993); Том. 68, с. 116 (1990). ( Статья )
- ^ Стилле, Дж. К.; Эчаваррен, AM; Уильямс, РМ; Хендрикс, Дж. А. Органический синтез , Сб. Том. 9, с. 553 (1998 год); Том. 71, с. 97 (1993). ( Статья )
- ^ Перейти обратно: а б с д и ж г час я дж к л Курти, Л.; Чако, Б. Стратегическое применение названных реакций в органическом синтезе ; Эльзевир: Берлингтон, 2005.
- ^ Митчелл, Теннесси Дж. Органомет. хим. , 1986 , 304 , 1–16.
- ^ Перейти обратно: а б Митчелл, TN Synthesis , 1992 , 803–815. ( два : 10.1055/s-1992-26230 )
- ^ Перейти обратно: а б Farina, V. Pure Appl. Chem. , 1996 , 68 , 73–78. ( дои : 10.1351/pac199668010073 ).
- ^ Перейти обратно: а б с д и ж г час я дж к л м н тот п Фарина, В.; Кришнамурти, В.; Скотт, WJ Реакция Стилле ; Вили: Онлайн, 2004. ( два : 10.1002/0471264180.or050.01 ).
- ^ Перейти обратно: а б с д и ж г час я Эспинет, П.; Эчаваррен, А. М. Анжью. хим. Межд. Эд. , 2004 , 43 , 4704–4734.( два : 10.1002/anie.200300638 )
- ^ Паттенден, Г.; Синклер, DJ Дж. Органомет. хим. , 2002 , 653, 261–268.
- ^ Kosugi, M.; Fugami, K. J. Organomet. Chem. , 2002 , 19, 10–16.
- ^ Перейти обратно: а б Пьер Жене, Ж.; Савиньяк, MJ Organomet. хим. , 1999 , 576, 305–317.
- ^ Кордова, К.; Варфоломей, К.; Мартинес-Илардуя, младший медик; Эспинет, P. ACS Catal ., 2015 , 5 , 3040–3053.( два : 10.1021/acscatal.5b00448 ).
- ^ Азарян, Д.; Дуа, СС; Иборн, К.; Уолтон, доктор медицинских наук Дж. Органомет. хим. , 1976 , 117 , С55-С57. ( дои : 10.1016/S0022-328X(00)91902-8 )
- ^ Kosugi, M.; Shimizu, Y.; Migita, T. Chem. Lett. , 1977 , 6 , 1423–1424. ( дои : 10.1246/кл.1977.1423 )
- ^ Перейти обратно: а б Косуги, М.; Сасазава, К.; Сикидзу, Ю.; Мигита, Т. Chem. Летт. , 1977 , 6 , 301–302. ( дои : 10.1246/кл.1977.301 )
- ^ Kosugi, M.; Shimizu, Y.; Migita, T. J. Organomet. Chem. , 1977 , 129 , C36-C38. ( дои : 10.1016/S0022-328X(00)92505-1 )
- ^ Мильштейн, Д.; Стилл, Дж. К. Журнал Американского химического общества , 1978 , 100 , 3636–3638. ( два : 10.1021/ja00479a077 )
- ^ Мильштейн, Д.; Стилл, Дж. К. Журнал Американского химического общества , 1979 , 101 , 4992–4998. ( два : 10.1021/ja00511a032 )
- ^ Мильштейн, Д.; Стилле, JK J. Org. хим. , 1979 , 44 , 1613–1618. ( два : 10.1021/jo01324a006 )
- ^ Женат, Алабама; Эспинет, П.; Gallego, AM J. Am, Soc. , 2000 , 122 , 11771-11782. ( два : 10.1021/ja001511o )
- ^ Перейти обратно: а б с д Крэбтри, Р. Х. Металлоорганическая химия переходных металлов , 5-е изд.; Уайли: Нью-Йорк, 2009.
- ^ Перейти обратно: а б Перес-Темпрано, Миннесота; Гальего, AM; Касарес, Х.А.; Эспинет, П. Металлоорганические соединения , 2011 , 30 , 611–617. ( два : 10.1021/om100978w ).
- ^ Перейти обратно: а б Миннити, Д. Неорг. Chem , 1994 , 33 , 2631–2634.( два : 10.1021/ic00090a025 ).
- ^ Перейти обратно: а б Касадо, Алабама; Эспинет, П. Металлоорганические соединения , 1998 , 17 , 954–959. ( два : 10.1021/om9709502 ).
- ^ Перейти обратно: а б Лэндис, ЧР; Фирман, Т.К. Рут, Д.М.; Кливленд, Т. Журнал Американского химического общества , 1998 , 120 , 1842–1854. ( дои : 10.1021/ja9710114 ).
- ^ Висенте, Дж.; Аркас, А.; Баутиста, Д. Металлоорганические соединения , 1997 , 16 , 2127–2138. ( два : 10.1021/om961094h ).
- ^ Перейти обратно: а б Пирсон, RG Inorg. Chem , 1973 , 12 , 712–713.( два : 10.1021/ic50121a052 ).
- ^ Перейти обратно: а б с Гарсия-Мельчор, М.; Брага, ААС; Льедос, А.; Ухаке, Г.; Масерас, F. Chem. , 2013 , 46 , 2626–2634. ( два : 10.1021/ar400080r )
- ^ Перейти обратно: а б с д Гилли, А.; Стилл, Дж. К. Журнал Американского химического общества , 1980 , 102 , 4933–4941. ( два : 10.1021/ja00535a018 ).
- ^ Перейти обратно: а б с д Браун, Дж. М.; Кули, Н. А. Хим. Ред. , 1988 , 88 , 1031–1046. ( два : 10.1021/cr00089a003 ).
- ^ МакКиллоп, А.; Абель, EW; Стоун, ФГА; Уилкинсон, Г. Комплексная металлоорганическая химия II , Elsevier Scientific: Оксфорд, 1995.
- ^ Перейти обратно: а б Фарина, В.; Журнал Американского химического общества , 1991 , 113 , 9585–9595. ( два : 10.1021/ja00025a025 ).
- ^ Перейти обратно: а б «Реакция Стилле» (PDF) . hwpi.harvard.edu .
- ^ Либескинд, LS; Фенгл, RW J. Org. , 1990 , 55 , 5359–5364. ( дои : 10.1021/jo00306a012 ).
- ^ Фарина, В.; Кападия, С.; Бришнан, Б.; Ван, К.; Либескинд, LS , Org. 1994 , J 59 , 5905–5911. ( дои : 10.1021/jo00099a018 ).
- ^ Перейти обратно: а б Ми, SPH; Ли, В.; Болдуин, Дж. Э. Энджью. хим. Межд. Эд. , 2004 , 43 , 1132–1136.
- ^ Либескинд, Л.С.; Пенья-Кабрера, Э. Органические синтезы , Сб. Полет. 10, с. 9 (2004 г.); Полет. 77, с. 135 (2000). ( Статья )
- ^ Скотт, WJ; Стилл, Дж. К. Журнал Американского химического общества , 1986 , 108 , 3033–3040. ( два : 10.1021/ja00271a037 ).
- ^ Джонсон, ЧР; Адамс, JP; Браун, член парламента; Сенанаяке, CBW Tetrahedron Lett ., 1992 , 33 , 919–922. ( два : 10.1016/S0040-4039(00)91576-4 )
- ^ Наир, В.; Тернер, Джорджия; Чемберлен, SD Журнал Американского химического общества , 1987 , 109 , 7223–7224. ( два : 10.1021/ja00257a071 ).
- ^ Жуссом, Б.; Квон, В.; Верлак, Дж.Б.; Денат, Ф.; Дубак, Дж. Синлетт , 1993 , 117–118. ( два : 10.1055/s-1993-22368 )
- ^ Шеффи, ФК; Годшалкс, JP; Стилл, Дж. К. Журнал Американского химического общества , 1984 , 106 , 4833–4840. ( два : 10.1021/ja00329a032 )
- ^ «Оловоорганические реагенты» .
- ^ Дитмар Зейферт (1959). «Ди -н -бутилдивинилолово». Орг. Синтез . 39 : 10. дои : 10.15227/orgsyn.039.0010 .
- ^ Вольф, Дж.; Реборс, RJ Org. хим. , 2003 , 68 7551–7554. ( дои : 10.1021/jo0347056 ).
- ^ Крисп, GT; Глинк, П.Т. Тетраэдр , 1994 , 50 , 2623. ( два : 10.1016/S0040-4020(01)86978-7 )
- ^ Бэйли, TR Tetrahedron Lett ., 1986 , 27 , 4407. ( два : 10.1016/S0040-4039(00)84964-3 ).
- ^ Бао, З.; Чан, В.; Ю, Л. Хим. Матер. , 1993 , 5 , 2–3. ( два : 10.1021/cm00025a001 ).
- ^ Бао, З.; Чан, ВК; Ю, Л. Журнал Американского химического общества , 1995 , 117 , 12426-12435. ( два : 10.1021/ja00155a007 ).
- ^ Солнце, СС; Льюис, Дж. Э.; Чжан, Дж.; Цзян, X.; Чжан, К.; Матос, Т.; Ли, Р.; Полим. хим. , 2010 , 1 , 663–669. ( дои : 10.1039/B9PY00324J )
- ^ Лебсак, AD; Линк, Джей Ти; Оверман, Ле; Стернс, бакалавр наук, Журнал Американского химического общества , 2002 , 124 , 9008–9009. ( два : 10.1021/ja0267425 )
- ^ Массе, CE; Ян, М.; Соломон Дж.; Панек, Дж. С. Журнал Американского химического общества , 1998 , 120 , 4123–4134. ( дои : 10.1021/ja9743194 )
- ^ Мартин, Сан-Франциско; Хамфри, Дж. М.; Али, А.; Хиллер, MC Журнал Американского химического общества , 1999 , 121 , 866–867. ( дои : 10.1021/ja9829259 )
- ^ Кенде, AS; Кавамура, К.; ДеВита, Р.Дж. Журнал Американского химического общества , 1990 , 112 4070–4072. ( два : 10.1021/ja00166a072 ).
- ^ Кенде, А.С., Кох, К.; Дори, Г.; Калдор, И.; Лю, К. Журнал Американского химического общества , 1993 , 115 , 9842–9843. ( два : 10.1021/ja00074a078 ).
- ^ Хонг, К.Ю., Киши, Ю. Журнал Американского химического общества , 1991 , 113 , 9693–9694. ( два : 10.1021/ja00025a056 ).
- ^ Танимото, Н.; Герритц, Юго-Запад; Савабе, А.; Нода, Т.; Вилла, ЮАР; Масамунэ, С. Ангью. хим. Межд. Эд. , 2003 , 33 , 673–675. ( два : 10.1002/anie.199406731 ).
- ^ Эванс, Д.А.; Блэк, WC Журнал Американского химического общества , 1993 , 115 , 4497–4513. ( два : 10.1021/ja00064a011 ).
- ^ Тан, В.; Прусов, Е.В. Орг. Летт. , 2012 , 14 4690–4693. ( дои : 10.1021/ol302219x ).
- ^ Коулман, Р.С.; Вальчак, MC; Кэмпбелл, Э.Л. Журнал Американского химического общества , 2005 , 127 , 16036-16039. ( два : 10.1021/ja056217g ).
- ^ Рот, врач общей практики; Фарина, В.; Либескинд, Л.С.; Пенья-Кабрера, Э. Тетраэдр Летт. 1995 , 36 , 2191.
- ^ Ренальдо, AF; Лабади, JW; Стилле, Дж. К. «Органические синтезы » , Coll. Том. 8, с. 268 (1993); Том. 67, с. 86 (1989). ( Статья )
- ^ Реакции Стилле с тетраалкилстаннанами и фенилтриалкилстаннанами в легкоплавких смесях сахара, мочевины и соли Джованни Императо, Рудольф Вазольд, Буркхард Кениг Advanced Synthesis & Catalise Volume 348, выпуск 15, страницы 2243–47, 2006 г. два : 10.1002/adsc.2006
- ^ П. Эспине, А. М. Эчаваррен (2004). «Механизмы реакции Стилле». Angewandte Chemie, международное издание . 43 (36): 4704–4734. дои : 10.1002/anie.200300638 . ПМИД 15366073 .
- ^ Найт, SD; Оверман, Ле; Пайродо, Дж. Журнал Американского химического общества , 1993 , 115 , 9293–9294. ( два : 10.1021/ja00073a057 )
- ^ Monera, E.; Ortar, G. Biorg. Med. Chem. Lett. , 2000 , 10 , 1815–1818. ( два : 10.1016/S0960-894X(00)00344-9 ).
- ^ Дьеркос, AC; Стилле, Дж. К.; Хегедус, Л.С. Журнал Американского химического общества , 1990 , 112 , 8465–8472. ( два : 10.1021/ja00179a035 ).
- ^ Юэ, WS; Ли, JJ Org. Летт. , 2002 , 4 , 2201–2203. ( дои : 10.1021/ol0260425 )
Внешние ссылки
[ редактировать ]- Раздаточный материал с реакциями Стилле от группы Майерс.
- Реакция Стилле на сайте органической химии.org