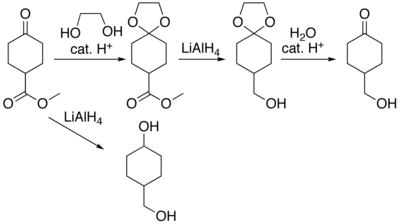
Во многих препаратах деликатных органических соединений определенные части молекул не выдерживают воздействия необходимых реагентов или химической среды. Эти части (функциональные группы) должны быть защищены . Например, литийалюминийгидрид — высокореактивный реагент, который эффективно восстанавливает сложные эфиры до спиртов . Он всегда реагирует с карбонильными группами, и его нельзя отпугивать никакими средствами. Когда сложный эфир необходимо восстановить в присутствии карбонила, необходимо предотвратить гидридную атаку карбонила. Один из способов сделать это — превратить карбонил в ацеталь , который не реагирует с гидридами. Ацеталь тогда называют защитной группой для карбонила. После завершения стадии гидрида водная кислота удаляет ацеталь, восстанавливая карбонил. Этот шаг называется снятием защиты .
Как правило, введение защитной группы является простым. Трудности, честно говоря, заключаются в их стабильности и выборочном удалении. Очевидные проблемы в стратегиях синтеза с защитными группами редко документируются в научной литературе. [2]
Ортогональная защита L-тирозина (Защитные группы отмечены синим цветом , аминокислота показана черным ). ( 1 ) Fmoc-защищенная аминогруппа , ( 2 , защищенная бензиловым эфиром ) карбоксильная группа , и ( 3 ) защищенная трет- бутиловым эфиром. фенольная гидроксильная группа тирозина,
Ортогональная защита — это стратегия, позволяющая специфическое снятие защиты с одной защитной группы в структуре с многократной защитой. Например, аминокислота тирозин может быть защищена как бензиловый эфир по карбоксильной группе, флуоренилметиленоксикарбамат по аминогруппе и трет- бутиловый эфир по фенольной группе. Бензиловый эфир можно удалить гидрогенолизом, флуоренилметиленокси-группу (Fmoc) - основаниями (такими как пиперидин), а фенольный трет -бутиловый эфир расщепляется кислотами (например, трифторуксусной кислотой).
Типичным примером этого применения является синтез пептидов Fmoc, при котором пептиды выращиваются в растворе и на твердой фазе. [3] Защитные группы в твердофазном синтезе в отношении условий реакции, таких как время реакции, температура и реагенты, могут быть стандартизированы так, что их можно проводить с помощью машины, при этом можно достичь выхода более 99%. В противном случае разделение образовавшейся смеси продуктов реакции практически невозможно (см. также § Промышленное применение ). [4]
Принципиальная схема твердотельного синтеза пептидов с ортогональными защитными группами X и Y.
Синтез твердотельных пептидов Fmoc с ортогональными защитными группами
Еще один важный пример ортогональных защитных групп встречается в химии углеводов . Поскольку углеводы и гидроксильные группы обладают очень похожей реакционной способностью, для успешного синтеза должна быть возможна трансформация, которая защищает или снимает защиту одной гидроксигруппы.
Различные группы расщепляются в кислотных или основных условиях, но остальные более необычны.
Ионы фтора образуют очень прочные связи с кремнием ; таким образом, защитные группы кремния почти всегда удаляются ионами фтора. Каждый тип противоиона, то есть реагента для расщепления, также может избирательно расщеплять различные защитные группы кремния в зависимости от стерических затруднений . Преимущество лабильных к фториду защитных групп заключается в том, что никакая другая защитная группа не подвергается воздействию условий расщепления.
Липазы и другие ферменты расщепляют эфиры при биологическом pH (5–9) и температуре (30–40 °С). Поскольку ферменты обладают очень высокой субстратной специфичностью, метод довольно редок, но чрезвычайно привлекателен.
Фотолабильные защитные группы несут хромофор , который активируется излучением соответствующей длины волны и поэтому может быть удален. [6] Для примера о здесь следует указать -нитробензильную группу.
Редкая двухслойная защитная группа представляет собой защищенную защитную группу, которая демонстрирует высокую стабильность.
Наиболее важными эфирами с общим использованием защитных групп являются ацетатные , бензоатные и пивалатные эфиры , поскольку они демонстрируют дифференцированное удаление. Стерически затрудненные эфиры менее подвержены нуклеофильной атаке:
Алифатические метиловые эфиры расщепляются с трудом и только в суровых условиях, поэтому их обычно используют только с хиноновыми фенолами. Однако полуацетали и ацетали расщепляются гораздо легче.
Эфиры триизопропилсилила (TIPS) — условия аналогичны TBS, но более длительное время реакции. [18]
трет- бутилдифенилсилил (TBDPS) — условия аналогичны TBS, но еще более длительное время реакции (в 100–250 раз медленнее, чем TBS, и в 5–10 раз медленнее, чем TIPS).
p , m — Диметоксибензиловый эфир — удаляется окислением DDQ или хлоридом церия и аммония. [24]
Ацетали:
Диметокситритил , [бис-(4-метоксифенил)фенилметил] (ДМТ) — Удаляется слабой кислотой. Группа ДМТ широко используется для защиты 5'-гидроксигруппы в нуклеозидах, особенно при синтезе олигонуклеотидов .
Метокситритил [(4-метоксифенил)дифенилметил] (ММТ) – удаляется кислотой и гидрогенолизом.
Бензилоксиметил — по стабильности сравним с MOM, MEM и SEM. [25] но допускает и восстановительное удаление: натрия в жидком аммиаке, [26] [27] каталитическое гидрирование (гидроксид палладия на активированном угле) или никель Ренея в этаноле [28] [29]
Этоксиэтиловые эфиры (ЭЭ) – расщепление более тривиально, чем у простых эфиров, например, 1 н. соляной кислоты. [30]
Аллил — удаляется трет- бутоксидом калия. [43] DABCO в метаноле, палладии на активированном угле или различных комплексах платины – в сочетании с кислотной обработкой. [44]
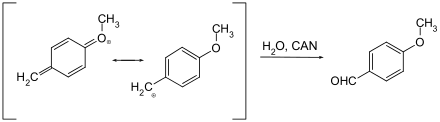
Метиловые эфиры. Расщепление осуществляется методом ТМСИ в дихлорметане, ацетонитриле или хлороформе. Альтернативный метод расщепления метиловых эфиров - BBr 3 в DCM.
Исключительный случай представляет собой бензилидензащитную группу, которая также допускает восстановительное расщепление. Это происходит либо посредством каталитического гидрирования, либо с использованием донора гидрида диизобутилалюминийгидрида (DIBAL). Расщепление с помощью DIBAL снимает защиту с одной спиртовой группы, поскольку бензильная группа остается в виде бензилового эфира на второй, пространственно затрудненной гидроксильной группе. [45] [46]
Амины имеют особое значение в синтезе пептидов , но являются весьма сильными нуклеофилами , а также относительно сильными основаниями . Эти характеристики подразумевают, что новые защитные группы для аминов постоянно находятся в стадии разработки. [47]
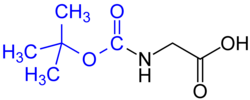
Аминные группы в первую очередь защищаются посредством ацилирования , обычно в виде карбамата . Когда карбамат снимает защиту, он выделяет углекислый газ . Наиболее распространенными карбаматами являются трет -бутоксикарбонильные, бензоксикарбонильные, флуоренилметиленоксикарбонильные и аллилоксикарбонильные соединения.
Другие, более экзотические аминные протекторы — это фталимиды , которые допускают восстановительное расщепление. [48] и трифторацетамиды, которые легко гидролизуются в основании. Индолы , пирролы и имидазолы — да и любой азагетероцикл — допускают защиту в виде N -сульфониламидов, которые слишком устойчивы по отношению к алифатическим аминам. [49] N -бензилированные амины можно удалить каталитическим гидрированием или восстановлением по Берчу, но они имеют явный недостаток по сравнению с карбаматами или амидами: они сохраняют основной азот.
Наиболее распространенными защитными группами карбонилов являются ацетали и обычно циклические ацетали с диолами. На втором месте также используются циклические ацетали с 1,2‑гидрокситиолами или дитиогликолями – так называемые O , S – или S , S –ацетали.
Ацетали можно удалить в кислых водных условиях. Для этих целей подходят минеральные кислоты. Ацетон — распространенный сорастворитель, используемый для ускорения растворения. Для некислотного метода расщепления используется комплекс хлорида палладия (II) с ацетонитрилом в ацетоне. [62] или хлорид железа(III) на силикагеле можно провести с обработкой в хлороформе. [63]
Циклические ацетали гораздо более устойчивы к кислотному гидролизу, чем ациклические ацетали. Следовательно, ациклические ацетали используются практически только тогда, когда требуется очень мягкое расщепление или когда при их высвобождении необходимо дифференцировать две разные защищенные карбонильные группы. [64]
Помимо O , O -ацеталей, S , O- и S , S -ацетали также имеют применение, хотя и скудное, в качестве защитных групп для карбонила. Тиолы , с которых начинаются эти ацетали, имеют очень неприятный запах и ядовиты, что сильно ограничивает их применение. Тиоацетали и смешанные S , O -ацетали, в отличие от чистых O , O -ацеталей, гораздо более устойчивы к кислотному гидролизу. Это позволяет селективно расщеплять последний в присутствии серозащищенных карбонильных групп.
Образование S , S -ацеталей обычно происходит аналогично образованию O , O -ацеталей при кислотном катализе из дитиола и карбонильного соединения. Из-за большей стабильности тиоацеталей равновесие лежит на стороне ацеталя. В отличие от случая О , О -ацеталя, для смещения равновесия не требуется удалять воду из реакционной смеси. [65]
S , O -ацетали гидролизуются в 10 000 раз быстрее, чем соответствующие S , S -ацетали. Их образование происходит аналогично тиоспирту. Также их расщепление протекает в аналогичных условиях и преимущественно через соединения ртути(II) во влажном ацетонитриле. [66]
Для альдегидов временная защита карбонильной группы - наличие кетонов в виде полуаминальных ионов показано ниже. Здесь применяется тот факт, что альдегиды представляют собой гораздо более активированные карбонилы, чем кетоны, и что многие реакции присоединения обратимы. [67] [68]
Важнейшими защитными группами карбоновых кислот являются эфиры различных спиртов. Иногда сложные эфиры защищают как ортоэфиры или оксазолины . [69]
Для спиртового компонента может быть достаточно многих групп, а конкретные условия расщепления, напротив, обычно весьма схожи: каждый сложный эфир может быть гидролизован в основном водно-спиртовом растворе. Вместо этого большинство защитных групп сложного эфира различаются по степени мягкости их образования из исходной кислоты.
Алкены редко нуждаются в защите или защищены. Они, как правило, участвуют только в нежелательных побочных реакциях электрофильной атаки, изомеризации или каталитической гидратации. Для алкенов в основном известны две защитные группы:
Защита посредством реакции Дильса-Альдера : превращение алкена в диен приводит к циклическому алкену, который, тем не менее, так же подвержен опасности электрофильной атаки, как и исходный алкен. Расщепление защитного диена протекает термически, поскольку реакция Дильса-Альдера является обратимой (равновесной) реакцией. [92] [93] [94]
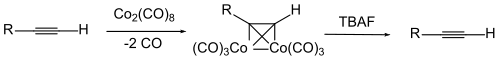
Для алкинов в любом случае существуют два типа защитных групп. Для концевых алкинов иногда важно замаскировать кислый атом водорода. Обычно это происходит в результате депротонирования (через сильное основание, такое как метилмагнийбромид или бутиллитий в тетрагидрофуране/ диметилсульфоксиде ) и последующей реакции с хлортриметилсиланом с образованием терминально защищенного ТМС алкина. [95] Расщепление происходит гидролитически – карбонатом калия в метаноле – или ионами фторида, например, фторидом тетрабутиламмония . [96]
Использование защитных групп широко распространено, но не без критики. [103] С практической точки зрения их использование добавляет к синтезу две стадии (последовательность защиты-снятия защиты), каждая из которых или обе могут значительно снизить химический выход . Важно отметить, что дополнительная сложность препятствует использованию синтетического полного синтеза при разработке лекарств . Напротив, биомиметический синтез не использует защитные группы. В качестве альтернативы Бэран представил новый синтез соединения гапалиндола U без защитных групп. Ранее опубликованный синтез [104] [105] [106] по словам Барана, содержал 20 шагов с множеством манипуляций с защитными группами (два подтвержденных):
Защищенный и незащищенный синтез морского алкалоида хапалиндола U.
Синтез Хидеаки Муратаке в 1990 году с использованием защитных групп тозила (показано синим цветом).
Хотя использование защитных групп не является предпочтительным в промышленном синтезе, они все еще используются в промышленных контекстах, например, сукралоза (подсластитель) или Roche синтез осельтамивира компанией (Тамифлю, противовирусный препарат).
Чтобы предотвратить окисление вторичных спиртов перманганатом калия , их защищают ацеталированием ацетоном , а затем снимают защиту после окисления первичных спиртов до карбоновых кислот. [107]
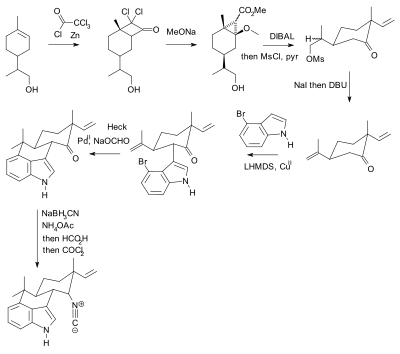
Очень ярким примером применения защитных групп в синтезе натуральных продуктов является полный синтез палитоксиновой кислоты в 1994 году исследовательской группой Ёсито Киши . [108] Здесь 42 функциональные группы (39 гидроксилов, один диол, аминогруппа и карбоновая кислота) нуждались в защите. Они проходили через 8 различных защитных групп (метиловый эфир, пять ацеталей, 20 эфиров TBDMS, девять п- метоксибензиловых эфиров, четыре бензоата, метилполуацеталь, ацетонацеталь и сложный эфир SEM). [109]
Палитоксин
Введение или модификация защитной группы иногда влияет на реакционную способность всей молекулы. Например, на схеме ниже представлен отрывок синтеза аналога Митомицина С Данишефского . [110]
Часть синтеза аналога Митомицина С с измененной реакционной способностью за счет замены защитной группы.
Обмен защитной группы с метилового эфира на МОМ-эфир ингибирует раскрытие эпоксида в альдегид .
Химия защитных групп находит важное применение в автоматизированном синтезе пептидов и нуклеозидов. Этот метод был введен в область синтеза пептидов Робертом Брюсом Меррифилдом в 1977 году. [111] Для синтеза пептидов с помощью автоматизированной машины используется ортогональность группы Fmoc (основное расщепление), трет -бутильной группы (кислотное расщепление) и различные защитные группы для функциональных групп на боковых цепях аминокислот. [112] используется до четырех различных защитных групп на одно азотистое основание Для автоматического синтеза последовательностей ДНК и РНК при синтезе олигонуклеотидов . Фактически процедура начинается с окислительно-восстановительной химии защищенного атома фосфора. Трехкоординированный фосфор, используемый из-за высокой реакционной способности, помечен цианоэтильной защитной группой на свободном кислороде. После стадии связывания следует окисление до фосфата, при котором защитная группа остается прикрепленной. Свободные ОН-группы, не прореагировавшие на стадии сочетания, ацетилируются на промежуточной стадии. Эти дополнительно введенные защитные группы затем препятствуют тому, чтобы эти ОН-группы могли соединиться в следующем цикле. [113]
^ Тод К. Джонс, Роберт А. Ример, Ричард Десмонд, Сандер Г. Миллс: «Химия трикарбонильных полукеталей и применение технологии Эванса для полного синтеза иммунодепрессанта (-)-FK-506», в: J. Am. хим. Соц. , 1990 , 112 , стр. 2998–3017; doi:10.1021/ja00164a023 .
^ Дитер Зеебах, Хак-Фун Чоу, Ричард Ф.В. Джексон, Мариус А. Саттер, Сувит Тайсривонгс, Юрг Циммерманн: «(+)-11,11 ′ -Ди-О-метилэлаиофилиден – препарат из элайофилина и полный синтез из (R) -3-гидроксибутират и (S)-малат», в: Liebigs Ann. хим. , 1986 , стр. 1281–1308; doi:10.1002/jlac.198619860714 .
^ Дэвид А. Эванс, Стивен В. Калдор, Тодд К. Джонс, Джон Кларди, Томас Дж. Стаут: «Полный синтез макролидного антибиотика цитоварицина», в: J. Am. хим. Соц. , 1990 , 112 , стр. 7001–7031; doi:10.1021/ja00175a038 .
^ Джеймс А. Маршалл, Ричард Седрани: «Конвергентный, высокостереоселективный синтез субъединицы C-11-C-21 макбецинов», в: J. Org. хим. , 1991 , 56 , стр. 5496–5498; дои:10.1021/jo00019a004 .
^ Моррис Дж. Робинс, Висенте Самано, Марк Д. Джонсон: «Соединения, родственные нуклеиновой кислоте. 58. Окисление периодинана, селективное первичное снятие защиты и удивительно стереоселективное восстановление рибонуклеозидов, защищенных трет-бутилдиметилсилилом. Синтез 9-(β-D) -ксилофуранозил)аденин или 3'-дейтериоаденозин из аденозина», в: J. Org. хим. , 1990 , 55 , стр. 410–412; дои:10.1021/jo00289a004 .
^ Р. Роджер Ф. Ньютон, Дерек П. Рейнольдс, Колин Ф. Уэбб, Стэнли М. Робертс: «Краткий и эффективный полный синтез (±) метилового эфира простагландина D 2 с использованием нового метода расщепления диметил- т-бутилсилиловый эфир», в: J. Chem. Soc., Перкин Транс. 1 , 1981 , стр. 2055–2058; дои:10.1039/P19810002055 .
^ М. Л. Гарсия, Х. Паскуаль, Л. Боррас, Х. А. Андреу, Э. Фос, Д. Молеон, Г. Карганико, Ф. Аркамоне: «Синтез новых эфирных глицерофосфолипидов, структурно связанных с модулятором», в: Tetrahedron , 1991 , 47. , стр. 10023–10034; doi:10.1016/S0040-4020(01)96051-X .
^ Х. Нагаока, В. Рутч, Г. Шмидт, Х. Ито, М. Р. Джонсон, Ю. Киши: «Полный синтез рифамицинов. 1. Стереоконтролируемый синтез алифатического строительного блока», в: J. Am. хим. Соц. , 1980 , 102 , стр. 7962–7965; doi:10.1021/ja00547a037 .
^ Пол А. Вендер, Карлос Р.Д. Коррейя: «Внутримолекулярные фотоиндуцированные диен-диеновые цианоприсоединения: селективный метод синтеза сложных восьмичленных колец и полихинанов», в: J. Am. хим. Соц. , 1987 , 109 , стр. 2523–2525; doi:10.1021/ja00242a053 .
^ Карел Ф. Бернади, М. Браунер Флойд, Джон Ф. Полетто, Мартин Дж. Вайс: «Простагландины и их родственные соединения. 20. Синтез простагландинов посредством конъюгатного добавления реагентов транс-1-алкенилтриалкилаланат лития. Новый реагент для конъюгата 1, 4-приложения», в: J. Org. хим. , 1979 , 44 , стр. 1438–1447; дои:10.1021/jo01323a017 .
^ Элиас Дж. Кори, Харуки Нива, Йохен Нолле: «Полный синтез (S)-12-гидрокси-5,8,14-цис,-10-транс-эйкозатетраеновой кислоты (HETE Самуэльссона)», в: J. Am . хим. Соц. , 1978 , 100 , стр. 1942–1943; doi:10.1021/ja00474a058 .
^ Роберт К. Гадвуд, Рене М. Летт, Джейн Э. Виссинджер: «Полный синтез (±)-поитедиола и (±) 4-эпипоитедиола», в: J. Am. хим. Соц. , 1984 , 106 , стр. 3869–3870; дои:10.1021/ja00325a032 .
^ Джозеф П. Марино, Скотт Л. Дакс: «Эффективный метод десилилирования для получения о-хинонметидов: применение к синтезу (+)- и (-)-гексагидроканнабинола», в: J. Org. хим. , 1984 , 49 , стр. 3671–3672; дои:10.1021/jo00193a051 .
^ Джеймс А. Маршалл, Джозеф Д. Трометр, Брюс Э. Блау, Томас Д. Крут: «Стереохимия добавлений SN2 к ациклическим винилоксиранам», в J. Org. хим. , 1988 , 53 , стр. 4274–4282 doi:10.1021/jo00253a020 .
^ Роберт М. Уильямс, Питер Дж. Синклер, Дунгуань Чжай, Даймо Чен: «Практический асимметричный синтез α-аминокислот посредством конструкций углерод-углеродных связей на электрофильных глициновых матрицах», в: J. Am. хим. Соц. , 1988 , 110 , с. 1547–1557; дои:10.1021/ja00213a031 .
^ Гленн Л. Шталь, Родерих Уолтер, Кларк В. Смит: «Общая процедура синтеза моно-N-ацилированных 1,6-диаминогексанов», в: J. Org. хим. , 1978 , 43 , стр. 2285–2286; doi:10.1021/jo00405a045 .
^ Венг К. Чан, Питер Д. Уайт: Твердофазный пептидный синтез Fmoc , стр. 27–30.
^ Грегг Б. Филдс: Методы удаления группы Fmoc. (PDF; 663 КБ) В: Майкл В. Пеннингтон, Бен М. Данн (ред.): Протоколы синтеза пептидов, том 35, 1995 г., ISBN 978-0-89603-273-6, стр. 17–27.
^ Б. Либе, Х. Кунц: Твердофазный синтез опухолеассоциированного гликопептида антигена сиалил-Tn с частичной последовательностью «тандемного повтора» муцина MUC-1. В: Angew. Chem. том 109, 1997, стр. 629–631 (на немецком языке).
^ Мусса, Зиад; Д. Ромо (2006). «Мягкое снятие защиты с первичных N-(п-толуолсульфонил)амидов с помощью SmI 2 после трифторацетилирования». Синлетт . 2006 (19): 3294–3298. дои : 10.1055/s-2006-951530 .
^ Кван Су Ким, Ян Хон Сон, Бонг Хо Ли, Чи Сун Хан: «Эффективное и селективное расщепление ацеталей и кеталей с использованием хлорида железа, адсорбированного на силикагеле», в: J. Org. хим. , 1986 , 51 , стр. 404–407; дои:10.1021/jo00353a027 .
^ Сэмюэл Дж. Данишефски, Натан Б. Мантло, Деннис С. Ямасита, Гейл. Шульте: «Краткий путь к серии калихемицин-эсперамицин: кристаллическая структура прототипа агликона», в: J. Am. хим. Соц. , 1988 , 110 , стр. 6890–6891; doi:10.1021/ja00228a051 .
^ Джон Н. Хазелтин, Мария Пас Кабал, Натан Б. Мантло, Нобухару Ивасава, Деннис С. Ямасита, Роберт С. Коулман, Сэмюэл Дж. Данишефски, Гейл К. Шульте: «Полный синтез калихеамицинона: новые механизмы для приведения в действие восстановительная циклоароматизация родственных агликонов», в: J. Am. хим. Соц. , 1991 , 113 , стр. 3850–3866; doi:10.1021/ja00010a030 .
^ Г. Бодуэн, Д. Бондон, Ю. Пьетрасанта, Б. Пуччи: «Реакции трансцетализации - II: Влияние стерических и электронных факторов на энергию кетализации», в: Tetrahedron , 1978 , 34 , стр. 3269–3274; дои: 10.1016/0040-4020(78)80243-9 .
^ Ахмед М. Тафеш, Йенс Вейгуни: «Обзор селективного каталитического восстановления ароматических нитросоединений в ароматические амины, изоцианаты, карбаматы и мочевины с использованием CO», в: Chem. Ред. , 1996 , 96 , стр. 2035–2052; дои:10.1021/cr950083f .
^ Эван Л. Оллред, Бойд Р. Бек, Кент Дж. Вурхис: «Образование двойных углерод-углеродных связей в результате реакции вицинальных дигалогенидов с натрием в аммиаке», в: J. Org. хим. , 1974 , 39 , стр. 1426–1427; doi:10.1021/jo00926a024 .
^ Тимоти С. Батчер, Фэн Чжоу, Майкл Р. Детти: «Дебромирование вик-дибромидов диорганотеллуридами. 1. Стереоселективность, относительные скорости и механистические последствия», в: J. Org. хим. , 1998 , 63 , стр. 169–176; дои: 10.1021/jo9713363 .
^ Синтетические исследования морских алкалоидов хапалиндолов. Часть I Полный синтез (±)-гапалиндолов J и M Tetrahedron , Том 46, Выпуск 18, 1990 г. , Страницы 6331–6342 Хидеаки Муратакэ и Мицутака Нацуме два : 10.1016/S0040-4020(01)96005-3
^ Синтетические исследования морских алкалоидов хапалиндолов. Часть 2. Алюмогидридное восстановление богатой электронами двойной углерод-углеродной связи, сопряженной с индольным ядром Тетраэдр , Том 46, Выпуск 18, 1990 , Страницы 6343–6350 Хидеаки Муратаке и Мицутака Нацуме два : 10.1016/S0040-4020(01)96006-5
^ Синтетические исследования морских алкалоидов хапалиндолов. Часть 3 Полный синтез (±)-гапалиндолов H и U Tetrahedron , Том 46, Выпуск 18, 1990 г. , Страницы 6351–6360 Хидеаки Муратаке, Харуми Кумагами и Мицутака Нацуме два : 10.1016/S0040-4020(01)96007-7
^ К.К. Николау, Э.Дж. Соренсен: Классика в полном синтезе: цели, стратегии, методы , VCH Verlagsgesellschaft mbH, Weinheim 1996, стр. 711–729, ISBN 3-527-29284-5.
^ Питер Г.М. Вутс, Теодора В. Грин: Защитные группы Грина в органическом синтезе , 4-е изд., John Wiley & Sons Inc., Хобокен, Нью-Джерси, стр. 10–13; ISBN 0-471-69754-0.
^ Дж. М. МакКлюр, Сэмюэл Дж. Данишефски: «Новая реакция арилирования Хека: быстрый доступ к аналогам FR 900482», в: J. Am. хим. Соц. , 1993 , 115 , стр. 6094–6100; doi:10.1021/ja00067a026 .
^ Меррифилд, РБ; Барани, Г.; Косанд, WL; Энгельхард, М.; Мойсов, С. (1977). «Материалы 5-го Американского симпозиума по пептидам». Биохимическое образование . 7 (4): 93–94. дои : 10.1016/0307-4412(79)90078-5 .
^ Венг К. Чан, Питер Д. Уайт: Твердофазный пептидный синтез Fmoc . Перепечатка 2004 г., Oxford University Press, ISBN 0-19-963724-5.
Филип Дж. Коценски : Группы защиты , 1-е изд., Георг Тиме Верлаг, Штутгарт 1994, ISBN 3-13-135601-4.
Питер Г.М. Вутс, Теодора В. Грин: Защитные группы Грина в органическом синтезе , 4-е изд., John Wiley & Sons Inc., Хобокен, Нью-Джерси, ISBN 0-471-69754-0.
Arc.Ask3.Ru Номер скриншота №: cc0be67e51194ea07f7871df90748548__1712693940 URL1:https://arc.ask3.ru/arc/aa/cc/48/cc0be67e51194ea07f7871df90748548.html Заголовок, (Title) документа по адресу, URL1: Protecting group - Wikipedia
Данный printscreen веб страницы (снимок веб страницы, скриншот веб страницы), визуально-программная копия документа расположенного по адресу URL1 и сохраненная в файл, имеет: квалифицированную, усовершенствованную (подтверждены: метки времени, валидность сертификата), открепленную ЭЦП (приложена к данному файлу), что может быть использовано для подтверждения содержания и факта существования документа в этот момент времени. Права на данный скриншот принадлежат администрации Ask3.ru, использование в качестве доказательства только с письменного разрешения правообладателя скриншота. Администрация Ask3.ru не несет ответственности за информацию размещенную на данном скриншоте. Права на прочие зарегистрированные элементы любого права, изображенные на снимках принадлежат их владельцам. Качество перевода предоставляется как есть. Любые претензии, иски не могут быть предъявлены. Если вы не согласны с любым пунктом перечисленным выше, вы не можете использовать данный сайт и информация размещенную на нем (сайте/странице), немедленно покиньте данный сайт. В случае нарушения любого пункта перечисленного выше, штраф 55! (Пятьдесят пять факториал, Денежную единицу (имеющую самостоятельную стоимость) можете выбрать самостоятельно, выплаичвается товарами в течение 7 дней с момента нарушения.)

 Принципиальная схема твердотельного синтеза пептидов с ортогональными защитными группами X и Y.
Принципиальная схема твердотельного синтеза пептидов с ортогональными защитными группами X и Y.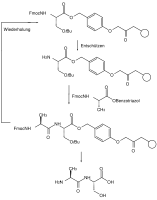 Синтез твердотельных пептидов Fmoc с ортогональными защитными группами
Синтез твердотельных пептидов Fmoc с ортогональными защитными группами