Реакция Дильса-Альдера
| Реакция Дильса-Альдера | |
|---|---|
| Тип реакции | Циклоприсоединение |
| Идентификаторы | |
| Портал органической химии | Реакция Дильса-Альдера |
| RSC Идентификатор онтологии | RXNO: 0000006 |
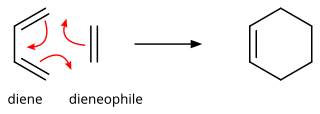
В органической химии реакция Дильса -Альдера представляет собой химическую реакцию между сопряженным диеном и замещенным алкеном , обычно называемым диенофилом , с образованием замещенного производного циклогексена . Это типичный пример перициклической реакции с согласованным механизмом . Более конкретно, оно классифицируется как термически разрешенное [4+2] циклоприсоединение с символом Вудворда – Хоффмана [ π 4 s + π 2 s ]. Впервые она была описана Отто Дильсом и Куртом Альдером в 1928 году. За открытие этой реакции они были удостоены Нобелевской премии по химии в 1950 году. Благодаря одновременному образованию двух новых связей углерод-углерод реакция Дильса-Альдера обеспечивает надежный способ формирования шестичленных колец с хорошим контролем над регио- и стереохимическими результатами. [1] [2] Следовательно, он послужил мощным и широко применяемым инструментом для усложнения химической структуры синтеза натуральных продуктов и новых материалов. [3] [4] Основная концепция также была применена к π-системам с участием гетероатомов , таких как карбонилы и имины , которые образуют соответствующие гетероциклы ; этот вариант известен как гетеро-реакция Дильса-Альдера . Реакция также была распространена на кольца других размеров, хотя ни одно из этих обобщений не соответствовало образованию шестичленных колец с точки зрения масштаба или универсальности. Из-за отрицательных значений ΔH ° и ΔS ° для типичной реакции Дильса-Альдера микроскопическое обращение реакции Дильса-Альдера становится благоприятным при высоких температурах, хотя это имеет синтетическое значение только для ограниченного диапазона реакций Дильса-Альдера. Аддукты ольхи, как правило, с некоторыми особыми структурными особенностями; эта обратная реакция известна как ретро-реакция Дильса-Альдера . [5]
Механизм
[ редактировать ]Реакция является примером согласованной перициклической реакции. [6] Считается, что это происходит через одно циклическое переходное состояние, [7] без образования промежуточных продуктов в ходе реакции. Таким образом, реакция Дильса-Альдера определяется соображениями орбитальной симметрии: она классифицируется как [ π 4 s + π 2 s ] циклоприсоединение, что указывает на то, что она протекает посредством супрафациального /супрафациального взаимодействия электронной системы 4π (диеновая структура ) с электронной системой 2π (диенофильная структура), взаимодействие, которое приводит к переходному состоянию без дополнительного энергетического барьера, налагаемого орбитальной симметрией, и позволяет относительно легко протекать реакции Дильса-Альдера. [8]
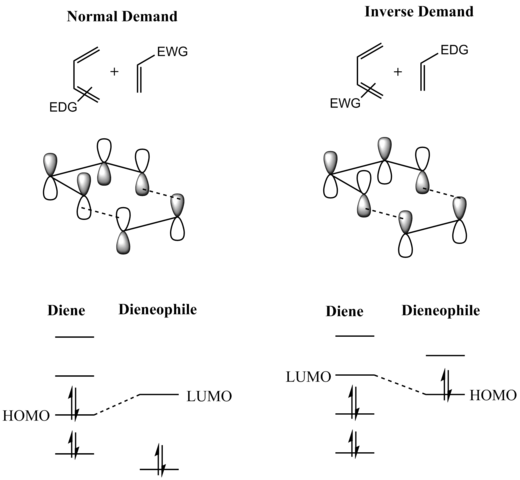
Рассмотрение граничных молекулярных орбиталей реагентов (FMO) объясняет, почему это так. (Тот же вывод можно сделать из диаграммы орбитальной корреляции или анализа Дьюара-Циммермана.) Для более распространенной «нормальной» реакции Дильса-Альдера с требованием электронов более важным из двух взаимодействий ВЗМО/НСМО является взаимодействие между электрон- богатого диена ψ 2 как самая высокая занятая молекулярная орбиталь (ВЗМО), а π * электронодефицитного диенофила как самая нижняя незанятая молекулярная орбиталь (НСМО). Однако энергетическая щель HOMO-LUMO достаточно близка, поэтому роли можно поменять местами, переключив электронные эффекты заместителей на два компонента. В обратной (обратной) реакции Дильса-Альдера с электронозапросом электроноакцепторные заместители на диене понижают энергию его пустой ψ 3 -орбитали, а электронодонорные заместители на диенофиле повышают энергию его заполненной π-орбитали настолько, что взаимодействие между этими двумя орбиталями становится наиболее энергетически значимым стабилизирующим орбитальным взаимодействием. Независимо от ситуации, ВЗМО и НСМО компонентов находятся в фазе, и в результате возникает связывающее взаимодействие, как можно видеть на диаграмме ниже. Поскольку реагенты находятся в основном состоянии, реакция инициируется термически и не требует активации светом. [8]
«Преобладающее мнение» [9] [10] [11] [12] заключается в том, что большинство реакций Дильса-Альдера протекают по согласованному механизму; однако этот вопрос тщательно оспаривается. Несмотря на то, что подавляющее большинство реакций Дильса-Альдера демонстрируют стереоспецифическое син-присоединение двух компонентов, было постулировано наличие бирадикального промежуточного соединения. [7] (и подтверждается вычислительными данными) на том основании, что наблюдаемая стереоспецифичность не исключает двухэтапного добавления с участием промежуточного продукта, который распадается до продукта быстрее, чем он может вращаться, что позволяет осуществить инверсию стереохимии.
Скорость заметно увеличивается, когда определенные реакции Дильса-Альдера проводятся в полярных органических растворителях, таких как диметилформамид и этиленгликоль . [13] и даже в воде. [14] реакция циклопентадиена и бутенона Например, в воде протекает в 700 раз быстрее по сравнению с 2,2,4-триметилпентаном в качестве растворителя. [14] Было предложено несколько объяснений этого эффекта, например, увеличение эффективной концентрации за счет гидрофобной упаковки. [15] или стабилизация переходного состояния водородными связями. [16]
Геометрия диенового и диенофильного компонентов распространяется на стереохимические детали продукта. В частности, для межмолекулярных реакций предпочтительное позиционное и стереохимическое соотношение заместителей двух компонентов по сравнению друг с другом контролируется электронными эффектами. Однако для внутримолекулярных реакций циклоприсоединения Дильса–Альдера конформационная устойчивость структуры переходного состояния может иметь подавляющее влияние.
Региоселективность
[ редактировать ]Теория пограничных молекулярных орбиталей также использовалась для объяснения закономерностей региоселективности, наблюдаемых в реакциях Дильса-Альдера замещенных систем. Расчет энергии и орбитальных коэффициентов граничных орбиталей компонентов [17] дает картину, которая хорошо согласуется с более простым анализом резонансных эффектов заместителей, как показано ниже.
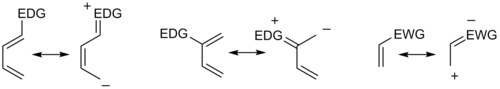
В целом, региоселективность, обнаруженная как для нормальной, так и для реакции Дильса-Альдера с обратным электронным требованием, соответствует так называемому правилу орто-пара , поскольку циклогексеновый продукт несет заместители в положениях, которые аналогичны орто- и пара -положениям дизамещенных аренов. Например, в сценарии нормального спроса диен, несущий электронодонорную группу (EDG) в C1, имеет самый большой коэффициент HOMO в C4, в то время как диенофил с электроноакцепторной группой (EWG) в C1 имеет самый большой коэффициент LUMO в C4. С2. Объединение этих двух коэффициентов дает «орто» произведение, как показано в случае 1 на рисунке ниже. Диен, замещенный в положении C2, как в случае 2 ниже, имеет наибольший коэффициент ВЗМО в положении C1, что приводит к образованию «пара»-продукта. Аналогичный анализ для соответствующих сценариев обратного спроса дает аналогичные продукты, как это видно в случаях 3 и 4. Исследуя приведенные выше канонические мезомерные формы, легко убедиться, что эти результаты соответствуют ожиданиям, основанным на учете электронной плотности и поляризация.

В целом, что касается наиболее энергетически согласованной пары HOMO-LUMO, максимизация энергии взаимодействия за счет образования связей между центрами с наибольшими граничными орбитальными коэффициентами позволяет предсказать основной региоизомер, который возникнет в результате данной комбинации диен-диенофил. [8] При более сложном лечении три типа заместителей ( Z отвод : понижение ВЗМО и НСМО (CF 3 , NO 2 , CN, C(O)CH 3 ), X отдача : повышение ВЗМО и НСМО (Me, OMe, NMe 2 ) , C- конъюгирование : рассматривается повышение HOMO и понижение LUMO (Ph, винил)), в результате чего получается в общей сложности 18 возможных комбинаций. Максимизация орбитального взаимодействия правильно предсказывает продукт во всех случаях, для которых доступны экспериментальные данные. Например, в редких комбинациях, включающих группы X как на диене, так и на диенофиле, может отдаваться предпочтение паттерну 1,3-замещения, но этот результат не объясняется аргументом упрощенной резонансной структуры. [18] Однако случаи, когда резонансный аргумент и совпадение наибольших орбитальных коэффициентов не совпадают, редки.
Стереоспецифичность и стереоселективность
[ редактировать ]Реакции Дильса-Альдера, как согласованные циклоприсоединения, стереоспецифичны . Стереохимическая информация о диене и диенофиле сохраняется в продукте в виде син- присоединения по отношению к каждому компоненту. Например, заместители в цис- ( транс -соответственном) отношении двойной связи диенофила приводят к появлению заместителей, которые являются цис- ( транс -соответственно) у тех же атомов углерода по отношению к циклогексеновому кольцу. Аналогично, цис , цис- и транс , транс -дизамещенные диены дают цис- заместители у этих атомов углерода продукта, тогда как цис , транс -дизамещенные диены дают транс -заместители: [19] [20]


Реакции Дильса-Альдера, в которых соседние стереоцентры генерируются на двух концах вновь образованных одинарных связей, предполагают два различных возможных стереохимических результата. Это стереоселективная ситуация, основанная на относительной ориентации двух отдельных компонентов, когда они реагируют друг с другом. В контексте реакции Дильса-Альдера переходное состояние, в котором наиболее значимый заместитель (электроноакцепторная и/или конъюгирующая группа) в диенофиле ориентирован к диеновой π-системе и проскальзывает под ней по мере протекания реакции, представляет собой известное как эндопереходное состояние. В альтернативном экзопереходном состоянии он ориентирован от него. используются более широко (В стереохимической номенклатуре термины эндо и экзо .)
В случаях, когда диенофил имеет один электроноакцепторный/сопряжающий заместитель или два электроноакцепторных/сопряжающих заместителя, цис-по отношению друг к другу, результат часто можно предсказать. В этих сценариях Дильса-Альдера с «нормальным спросом» эндопереходное состояние обычно предпочтительнее, несмотря на то, что оно часто более стерически перегружено. Это предпочтение известно как эндо-правило Ольдера . Как первоначально заявил Альдер, предпочтительным является переходное состояние с «максимальным накоплением двойных связей». Эндоселективность обычно выше для жестких диенофилов, таких как малеиновый ангидрид и бензохинон ; для других, таких как акрилаты и кротонаты , селективность не очень выражена. [21]
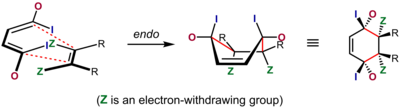
Наиболее широко распространенное объяснение происхождения этого эффекта - благоприятное взаимодействие между π-системами диенофила и диена, взаимодействие, описываемое как вторичный орбитальный эффект , хотя диполярное притяжение и притяжение Ван-дер-Ваальса также могут играть свою роль, и растворитель иногда может существенно повлиять на селективность. [6] [22] [23] Объяснение вторичного перекрытия орбит было впервые предложено Вудвордом и Хоффманном. [24] В этом объяснении орбитали, связанные с группой, находящейся в сопряжении с диенофильной двойной связью, перекрываются с внутренними орбиталями диена, что возможно только для эндопереходного состояния. Хотя первоначальное объяснение касалось только орбитали атома α по отношению к двойной связи диенофила, Салем и Хоук впоследствии предположили, что орбитали атома α и β участвуют в этом, когда позволяет молекулярная геометрия. [25]

Часто, как и в случае с сильнозамещенными диенами, очень объемистыми диенофилами или обратимыми реакциями (как в случае фурана в виде диена), стерические эффекты могут преобладать над нормальной эндоселективностью в пользу экзо- изомера.
Диен
[ редактировать ]Диеновый . компонент реакции Дильса-Альдера может быть как с открытой цепью, так и циклическим, и он может содержать множество различных типов заместителей [6] Однако он должен иметь возможность существовать в s- сцис -конформации, поскольку это единственный конформер, который может участвовать в реакции. Хотя бутадиены обычно более стабильны в s- транс -конформации, в большинстве случаев разница в энергии невелика (~ 2–5 ккал/моль). [26]
Объемный заместитель в положении C2 или C3 может увеличить скорость реакции, дестабилизируя конформацию s- транс и переводя диен в реакционноспособную конформацию s- сцис . 2- трет -бутилбута-1,3-диен в 27 раз более реакционноспособен, чем простой бутадиен. Например, [6] [27] И наоборот, диен, имеющий объемные заместители как у C2, так и у C3, менее реакционноспособен, поскольку стерические взаимодействия между заместителями дестабилизируют конформацию s- цис . [27]
Диены с объемными концевыми заместителями (С1 и С4) снижают скорость реакции, предположительно затрудняя сближение диена и диенофила. [28]
Особенно реакционноспособным диеном является 1-метокси-3-триметилсилоксибута-1,3-диен, иначе известный как диен Данишефского . [29] Он имеет особое синтетическое применение в качестве средства получения α,β-ненасыщенных циклогексеноновых систем путем удаления 1-метоксизаместителя после снятия защиты с енолсилилового эфира. Другие синтетически полезные производные диена Данишефского включают 1,3-алкокси-1-триметилсилокси-1,3-бутадиены (диены Брассара). [30] и 1-диалкиламино-3-триметилсилокси-1,3-бутадиены (диены Раваля). [31] Повышенная реакционная способность этих и подобных диенов является результатом синергического вклада групп доноров на C1 и C3, что значительно повышает ВЗМО по сравнению с сопоставимым монозамещенным диеном. [3]

Нестабильные (и, следовательно, высокореакционноспособные) диены могут быть полезны в синтетическом отношении, например, о - хинодиметаны могут быть получены in situ. Напротив, стабильные диены, такие как нафталин , требуют вынуждающих условий и/или высокореакционноспособных диенофилов, таких как N -фенилмалеимид . Антрацен , будучи менее ароматным (и, следовательно, более реакционноспособным для синтеза Дильса-Альдера), в своем центральном кольце может образовывать аддукт 9,10 с малеиновым ангидридом при 80 ° C и даже с ацетиленом , слабым диенофилом, при 250 ° C. [32]
Диенофил
[ редактировать ]В обычной реакции Дильса-Альдера диенофил имеет электроноакцепторную группу в сопряжении с алкеном; в сценарии обратного спроса диенофил конъюгируется с электронодонорной группой. [9] Диенофилы могут быть выбраны так, чтобы они содержали «замаскированную функциональность». Диенофил вступает в реакцию Дильса-Альдера с диеном, придающим такую функциональность молекуле продукта. Затем следует серия реакций для преобразования функциональной группы в желаемую группу. Конечный продукт не может быть получен за одну стадию DA, поскольку эквивалентный диенофил либо нереактивен, либо недоступен. Примером такого подхода является использование α-хлоракрилонитрила (CH 2 =CClCN). При реакции с диеном этот диенофил привносит в молекулу продукта функциональную группу α-хлорнитрила. Это «замаскированная функциональность», которую затем можно гидролизовать с образованием кетона . Диенофил α-хлоракрилонитрила является эквивалентом диенофила кетена (CH 2 =C=O), который дает тот же продукт за одну стадию DA. Проблема в том, что сам кетен не может быть использован в реакциях Дильса-Альдера, поскольку он реагирует с диенами нежелательным образом (путем [2+2]-циклоприсоединения), и поэтому необходимо использовать подход «маскированной функциональности». [33] Другими такими функциональными группами являются фосфониевые заместители (образующие экзоциклические двойные связи после реакции Виттига ), различные сульфоксидные и сульфонильные функциональные группы (оба являются ацетиленовыми эквивалентами) и нитрогруппы (кетеновые эквиваленты). [6]
Варианты классической реакции Дильса – Альдера.
[ редактировать ]Гетеро-Дильс-Альдер
[ редактировать ]Реакции Дильса-Альдера с участием хотя бы одного гетероатома также известны и называются гетеро-реакциями Дильса-Альдера. [34] Карбонильные группы , например, могут успешно реагировать с диенами с образованием дигидропирановых колец (реакция, известная как оксо-реакция Дильса-Альдера) , а имины могут использоваться либо в качестве диенофилов, либо в различных местах диена для образования различных N -гетероциклические соединения по реакции аза-Дильса-Альдера . Нитрозосоединения (RN=O) могут реагировать с диенами с образованием оксазинов . Хлоросульфонилизоцианат можно использовать в качестве диенофила для получения лактама Винса . [6] [35]
Активация кислоты Льюиса
[ редактировать ]Кислоты Льюиса , такие как хлорид цинка , трифторид бора , тетрахлорид олова или хлорид алюминия , могут катализировать реакции Дильса-Альдера путем связывания с диенофилом. Традиционно повышенная реакционная способность Дильса-Альдера приписывается способности кислоты Льюиса снижать LUMO активированного диенофила, что приводит к меньшему нормальному потреблению электронов орбитальной энергетической щели HOMO-LUMO и, следовательно, к большей стабилизации орбитальных взаимодействий. [36] [37] [38]
Однако недавние исследования показали, что это обоснование реакций Дильса-Альдера, катализируемых кислотой Льюиса, неверно. [39] [40] [41] [42] Обнаружено, что кислоты Льюиса ускоряют реакцию Дильса-Альдера за счет уменьшения дестабилизирующего стерического отталкивания Паули между взаимодействующими диеном и диенофилом, а не за счет снижения энергии LUMO диенофила и, следовательно, усиления нормального орбитального взаимодействия электронов. Кислота Льюиса связывается с диенофилом посредством донорно-акцепторного взаимодействия и посредством этого механизма поляризует плотность занятых орбиталей от реакционноспособной двойной связи C=C диенофила в сторону кислоты Льюиса. Эта уменьшенная плотность занятых орбиталей двойной связи C=C диенофила, в свою очередь, будет участвовать в менее отталкивающем орбитальном взаимодействии «закрытая оболочка-закрытая оболочка» с входящим диеном, уменьшая дестабилизирующее стерическое отталкивание Паули и, следовательно, снижая Дильс- Реакционный барьер ольхи. Кроме того, катализатор-кислота Льюиса также увеличивает асинхронность реакции Дильса-Альдера, делая асимметричной занятую π-орбиталь, расположенную на двойной связи C=C диенофила. В результате эта повышенная асинхронность приводит к дополнительному уменьшению дестабилизирующего стерического отталкивания Паули, а также к уменьшению давления на реагенты, заставляющего их деформироваться, другими словами, это уменьшает дестабилизирующую активационную деформацию (также известную как энергия искажения). [43] Этот рабочий каталитический механизм известен как катализ, понижающий Паули . [44] который участвует в различных органических реакциях. [45] [46] [47]
Первоначальное обоснование реакций Дильса – Альдера, катализируемых кислотой Льюиса, неверно. [39] [48] [49] [50] потому что, помимо снижения энергии LUMO диенофила, кислота Льюиса также снижает энергию HOMO диенофила и, следовательно, увеличивает обратную потребность электронов в орбитальной энергетической щели LUMO-HOMO. Таким образом, действительно, катализаторы на основе кислот Льюиса усиливают нормальное орбитальное взаимодействие потребности электронов за счет снижения НСМО диенофила, но они одновременно ослабляют обратное орбитальное взаимодействие потребности электронов, также снижая энергию ВЗМО диенофила. Эти два противодействующих явления эффективно нейтрализуют друг друга, что приводит к почти неизменным орбитальным взаимодействиям по сравнению с соответствующими некатализируемыми реакциями Дильса-Альдера и делает это не активным механизмом реакций Дильса-Альдера, катализируемых кислотой Льюиса.
Асимметричный Дильс-Альдер
[ редактировать ]Для влияния на стереоселективность реакции Дильса-Альдера разработано множество методов, таких как использование хиральных вспомогательных веществ, катализ хиральными кислотами Льюиса , [51] и катализаторы из малых органических молекул . [6] оксазолидиноны Эванса , [52] оксазаборолидины , [53] [54] [55] бис -оксазолин -меди хелаты , [56] имидазолиновый катализ, [57] и существует множество других методологий проведения диастерео- и энантиоселективных реакций Дильса-Альдера.
Гексадегидро Дильса-Альдера
[ редактировать ]В реакции гексадегидро Дильса-Альдера вместо алкины и алкенов и диенов используются диины, образуя нестабильный промежуточный бензол , который затем можно улавливать с образованием ароматического продукта. Эта реакция позволяет образовывать сильно функционализированные ароматические кольца за одну стадию. [58] [59]
Применение и естественное возникновение
[ редактировать ]
Ретро-реакция Дильса-Альдера используется в промышленном производстве циклопентадиена . Циклопентадиен является предшественником различных норборненов , которые являются распространенными мономерами . Реакция Дильса-Альдера также используется для производства витамина B6 .
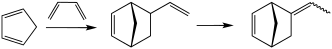
История
[ редактировать ]
Работа Дильса и Альдера описана в серии из 28 статей, опубликованных в журналах Justus Liebigs Annalen der Chemie и Berichte der deutschen chemischen Gesellschaft с 1928 по 1937 год. Авторами первых 19 статей были Дильс и Альдер, а авторами последующих статей были Дильса и других соавторов. [62] [63]
Приложения в полном синтезе
[ редактировать ]Реакция Дильса-Альдера была одним из этапов раннего получения стероидов кортизона и холестерина . [64] Реакция включала присоединение бутадиена к хинону.

Реакции Дильса-Альдера были использованы в оригинальном синтезе простагландинов F2α и E2 . [65] Реакция Дильса-Альдера устанавливает относительную стереохимию трех смежных стереоцентров на циклопентановом ядре простагландина. активация кислым тетрафторборатом меди Требовалась Льюиса.

Реакцию Дильса-Альдера использовали при синтезе префената динатрия . [66] биосинтетический предшественник аминокислот фенилаланина и тирозина .
При синтезе резерпина используется реакция Дильса-Альдера для установления цис -декалинового каркаса колец D и E. [67]

В другом синтезе резерпина цис -конденсированные кольца D и E были образованы реакцией Дильса-Альдера. Внутримолекулярный метод Дильса-Альдера пиранона, представленного ниже, с последующей экструзией диоксида углерода через ретро [4+2] дал бициклический лактам . Эпоксидирование с менее затрудненной α-границы с последующим раскрытием эпоксида по менее затрудненной стороне C18 обеспечивало желаемую стереохимию в этих положениях, тогда как цис -слияние достигалось с помощью гидрирования, снова происходящего в основном с менее затрудненной стороны. [68]

Пиранон аналогичным образом использовался в качестве диенофила при полном синтезе таксола . [69] Межмолекулярная реакция гидроксипирона и α,β-ненасыщенного эфира, показанная ниже, имела плохой выход и региоселективность; однако под действием фенилборной кислоты [70] желаемый аддукт может быть получен с выходом 61% после бороната неопентилгликолем расщепления . Стереоспецифичность реакции Дильса-Альдера в этом случае позволила определить четыре стереоцентра, которые перешли в конечный продукт.

Реакция Дильса-Альдера является ключевым этапом в синтезе (-)-фурахиноцина C. [71]

Таберсонин был получен реакцией Дильса-Альдера для установления относительной цис -стереохимии ядра алкалоида. Превращение цис -альдегида в соответствующий алкен путем олефинирования по Виттигу и последующий метатезис с замыканием кольца с помощью катализатора Шрока дало второе кольцо алкалоидного ядра. Диен в этом случае примечателен как пример 1-амино-3-силоксибутадиена, также известного как диен Раваля. [72]

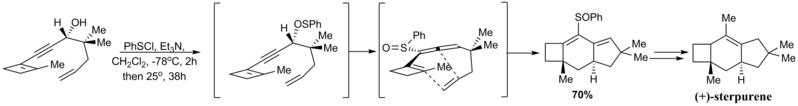
(+)-Стерпурен можно получить асимметричной реакцией DA. [73] который показал замечательную внутримолекулярную реакцию Дильса-Альдера с алленом . тиофенильной [2,3]-сигматропная перегруппировка группы с образованием сульфоксида, как показано ниже, протекала энантиоспецифично из-за заранее определенной стереохимии пропаргилового спирта. Таким образом, образовавшийся единственный алленовый изомер мог привести к тому, что реакция Дильса-Альдера протекала только на одной стороне образовавшегося «диена».
Тетрациклическое ядро антибиотика (-)-тетрациклина было получено реакцией Дильса-Альдера. Термически инициированное вращательное раскрытие бензоциклобутена привело к образованию о -хинодиметана, который вступал в межмолекулярную реакцию с образованием тетрациклинового скелета. Свободная гидроксильная группа диенофила является неотъемлемой частью успеха реакции, поскольку гидроксизащищенные варианты не вступали в реакцию в нескольких различных условиях реакции. [74]

Такемура и др. синтезировал кантаридин в 1980 году по реакции Дильса-Альдера с использованием высокого давления. [75]
Синтетические применения реакции Дильса-Альдера широко рассмотрены. [76] [77] [78] [79] [80]
См. также
[ редактировать ]- Циклоприсоединение Брэдшера
- Реакция Вагнера-Яурега
- Реакция Имина Дильса-Альдера
- Реакция Аза-Дильса-Альдера
Ссылки
[ редактировать ]- ^ Клёцель, MC (1948). «Реакция Дильса-Альдера с малеиновым ангидридом». Органические реакции . Том. 4. стр. 1–59. дои : 10.1002/0471264180.или004.01 . ISBN 978-0471264187 .
- ^ Холмс, HL (1948). «Реакция Дильса-Альдера, этиленовые и ацетиленовые диенофилы». Органические реакции . Том. 4. С. 60–173. дои : 10.1002/0471264180.или004.02 . ISBN 978-0471264187 .
- ^ Перейти обратно: а б Николау, КЦ; Снайдер, SA; Монтаньон, Т.; Вассиликояннакис, Г. (2002). «Реакция Дильса-Альдера в полном синтезе». Angewandte Chemie, международное издание . 41 (10): 1668–1698. doi : 10.1002/1521-3773(20020517)41:10<1668::AID-ANIE1668>3.0.CO;2-Z . ПМИД 19750686 .
- ^ Атилла Тасделен, Мехмет (2011). «Реакции щелчка Дильса-Альдера: последние применения в науке о полимерах и материалах». Полимерная химия . 2 (10): 2133–2145. дои : 10.1039/C1PY00041A .
- ^ Цвайфель, Г.С.; Нанц, Миннесота (2007). Современный органический синтез: Введение . WH Freeman and Co. ISBN 978-0-7167-7266-8 .
- ^ Перейти обратно: а б с д и ж г Кэри , Часть Б., стр. 474–526.
- ^ Перейти обратно: а б Дьюар, MJ; Оливелла, С.; Стюарт, Джей-Джей (1986). «Механизм реакции Дильса-Альдера: Реакции бутадиена с этиленом и цианоэтиленами». Журнал Американского химического общества . 108 (19): 5771–5779. дои : 10.1021/ja00279a018 . ПМИД 22175326 .
- ^ Перейти обратно: а б с Кэри , Часть А., стр. 836–50.
- ^ Перейти обратно: а б Кэри , Часть А., с. 839
- ^ Гаевски, Джей Джей; Петерсон, КБ; Кагель-младший (1987). «Изменение структуры переходного состояния в реакции Дильса-Альдера из-за эффектов вторичного кинетического изотопа дейтерия: реакция почти симметричного диена и диенофила почти синхронна». Журнал Американского химического общества . 109 (18): 5545–5546. дои : 10.1021/ja00252a052 .
- ^ Хоук, КН; Лин, Ю.Т.; Браун, ФК (1986). «Доказательства согласованного механизма реакции Дильса-Альдера бутадиена с этиленом». Журнал Американского химического общества . 108 (3): 554–556. дои : 10.1021/ja00263a059 . ПМИД 22175504 .
- ^ Гольдштейн, Э.; Бено, Б.; Хоук, КН (1996). «Прогнозирование относительных энергий и изотопных эффектов теорией функционала плотности для согласованных и ступенчатых механизмов реакции Дильса-Альдера бутадиена и этилена». Журнал Американского химического общества . 118 (25): 6036–6043. дои : 10.1021/ja9601494 .
- ^ Бреслоу, Р.; Го, Т. (1988). «Реакции Дильса-Альдера в неводных полярных растворителях. Кинетические эффекты хаотропных и антихаотропных агентов и β-циклодекстрина». Журнал Американского химического общества . 110 (17): 5613–5617. дои : 10.1021/ja00225a003 .
- ^ Перейти обратно: а б Райдаут, округ Колумбия; Бреслоу, Р. (1980). «Гидрофобное ускорение реакций Дильса-Альдера». Журнал Американского химического общества . 102 (26): 7816–7817. дои : 10.1021/ja00546a048 .
- ^ Бреслоу, Р.; Риццо, CJ (1991). «Хаотропные солевые эффекты в гидрофобно-ускоренной реакции Дильса-Альдера». Журнал Американского химического общества . 113 (11): 4340–4341. дои : 10.1021/ja00011a052 .
- ^ Блокзейл, Вильфрид; Энгбертс, Ян БФН (1992). «Влияние начального и переходного состояний на реакции Дильса-Альдера в воде и смешанных водных растворителях». Журнал Американского химического общества . 114 (13): 5440–5442. дои : 10.1021/ja00039a074 .
- ^ Эшби, ЕС; Чао, Л.-К.; Нойманн, HM (1973). «Механизмы металлоорганических реакций. XII. Механизм присоединения метилмагнийбромида к бензонитрилу». Журнал Американского химического общества . 95 (15): 4896–4904. дои : 10.1021/ja00796a022 .
- ^ Флеминг, И. (1990). Пограничные орбитальные и органохимические реакции . Чичестер, Великобритания: Wiley. ISBN 978-0471018193 .
- ^ Кирмсе, В.; Монч, Д. (1991). «Перегруппировки 1,4,4- и 2,2,5-триметилбицикло[3.2.1]окт-6-ильных катионов». Химические отчеты . 124 (1): 237–240. дои : 10.1002/cber.19911240136 .
- ^ Берубе, Г.; ДеЛонгшам, П. (1987). «Стереоселекция ациклического-1,5: синтез оптически активной боковой цепи витамина Е». Бюллетень Французского химического общества . 1 :103–115.
- ^ Хоук, КН; Лускус, ЖЖ (1971). «Влияние стерических взаимодействий на эндостереоселективность». Журнал Американского химического общества . 93 (18): 4606–4607. дои : 10.1021/ja00747a052 .
- ^ Кобуке, Ю.; Сугимото, Т.; Фурукава, Дж.; Фуэно, Т. (1972). «Роль притягивающих взаимодействий в эндо-экзо-стереоселективности реакций Дильса-Альдера». Журнал Американского химического общества . 94 (10): 3633–3635. дои : 10.1021/ja00765a066 .
- ^ Уильямсон, КЛ; Сюй, Ю.-Флорида (1970). «Стереохимия реакции Дильса-Альдера. II. Катализ кислотой Льюиса син-анти-изомерии». Журнал Американского химического общества . 92 (25): 7385–7389. дои : 10.1021/ja00728a022 .
- ^ Вудворд, РБ; Хоффманн, Р. (22 октября 2013 г.). Сохранение орбитальной симметрии . Вайнхайм. ISBN 9781483282046 . OCLC 915343522 .
{{cite book}}: CS1 maint: отсутствует местоположение издателя ( ссылка ) - ^ Ваннере, Чайтанья С.; Пол, Анкан; Эргес, Райнер; Хоук, КН; Шефер, Генри Ф.; Шлейер, Поль фон Раге (2007). «Существование вторичных орбитальных взаимодействий». Журнал вычислительной химии . 28 (1): 344–361. дои : 10.1002/jcc.20532 . ISSN 1096-987X . ПМИД 17109435 . S2CID 26096085 .
- ^ Кэри , Часть A, с. 149
- ^ Перейти обратно: а б Бэкер, HJ (1939). «2,3-Диттертиобутилбутадиен». Коллекция химических предприятий Нидерландов . 58 (7): 643–661. дои : 10.1002/recl.19390580712 .
- ^ Крейг, Д.; Шипман, Джей-Джей; Фаулер, РБ (1961). «Скорость реакции малеинового ангидрида с 1,3-диенами в зависимости от конформации диена». Журнал Американского химического общества . 83 (13): 2885–2891. дои : 10.1021/ja01474a023 .
- ^ Данишевский, С.; Китахара, Т. (1974). «Полезный диен для реакции Дильса-Альдера». Журнал Американского химического общества . 96 (25): 7807–7808. дои : 10.1021/ja00832a031 .
- ^ Савард, Дж.; Брассар, П. (1979). «Региоспецифический синтез хинонов с использованием винилкетенацеталей, полученных из ненасыщенных эфиров». Буквы тетраэдра . 20 (51): 4911–4914. дои : 10.1016/S0040-4039(01)86747-2 .
- ^ Козьмин, С.А.; Равал, В.Х. (1997). «Получение и реакционная способность Дильса-Альдера 1-амино-3-силокси-1,3-бутадиенов». Журнал органической химии . 62 (16): 5252–5253. дои : 10.1021/jo970438q .
- ^ Маргарет Аврам (1983). Органическая химия стр. 318-323. Издательство Академии Социалистической Республики Румыния
- ^ Ранганатан, С.; Ранганатан, Д.; Мехротра, АК (1977). «Кетеновые эквиваленты». Синтез . 1977 (5): 289–296. дои : 10.1055/s-1977-24362 . S2CID 260335918 .
- ^ Руш, WR (1991). «Внутримолекулярные реакции Дильса-Альдера». В Тросте, Б.М.; Флемминг, И. (ред.). Комплексный органический синтез . Том. 5. С. 513–550. дои : 10.1016/B978-0-08-052349-1.00131-1 . ISBN 978-0-08-052349-1 .
- ^ Греко, Пенсильвания; Ларсен, С.Д. (1990). «Реакции Дильса-Альдера на основе иминиевых ионов: N-бензил-2-азанорборен» (PDF) . Органические синтезы . 68 : 206. дои : 10.15227/orgsyn.068.0206 .
- ^ Хоук, Кендалл Н. (1 ноября 1975 г.). «Пограничная молекулярно-орбитальная теория реакций циклоприсоединения» . Отчеты о химических исследованиях . 8 (11): 361–369. дои : 10.1021/ar50095a001 . ISSN 0001-4842 .
- ^ Флеминг, Ян (2009). Молекулярные орбитали и органические химические реакции . Чичестер, Западный Суссекс, Великобритания: Wiley. ISBN 9780470746592 .
- ^ Клейден, Джонатан (2012). Органическая химия (2-е изд.). Оксфорд: Издательство Оксфордского университета. ISBN 9780199270293 .
- ^ Перейти обратно: а б Вермерен, Паскаль; Хэмлин, Тревор А.; Фернандес, Израиль; Бикельхаупт, Ф. Матиас (6 апреля 2020 г.). «Как кислоты Льюиса катализируют реакции Дильса-Альдера» . Angewandte Chemie, международное издание . 59 (15): 6201–6206. дои : 10.1002/anie.201914582 . ПМЦ 7187354 . ПМИД 31944503 .
- ^ Вермерен, Паскаль; Хэмлин, Тревор А.; Фернандес, Израиль; Бикельхаупт, Ф. Матиас (2020). «Происхождение повышения скорости и асинхронности в реакциях Дильса – Альдера, катализируемых иминием» . Химическая наука . 11 (31): 8105–8112. дои : 10.1039/D0SC02901G . ПМЦ 8163289 . ПМИД 34094173 .
- ^ Вермерен, Паскаль; Хэмлин, Тревор А.; Бикельхаупт, Ф. Матиас; Фернандес, Израиль (17 марта 2021 г.). «Бифункциональные реакции Дильса-Альдера, катализируемые донором водородной связи: происхождение стереоселективности и повышения скорости» . Химия: Европейский журнал . 27 (16): 5180–5190. дои : 10.1002/chem.202004496 . ПМК 8049058 . ПМИД 33169912 .
- ^ Вермерен, Паскаль; Тецца, Марко Далла; Донген, Мишель; Фернандес, Израиль; Бикельхаупт, Ф. Матиас; Хэмлин, Тревор А. (21 июля 2021 г.). «Реакции Дильса-Альдера, катализируемые кислотой Льюиса: тенденции реактивности в периодической таблице» . Химия: Европейский журнал . 27 (41): 10610–10620. дои : 10.1002/chem.202100522 . ПМК 8360170 . ПМИД 33780068 .
- ^ Вермерен, Паскаль; Хэмлин, Тревор А.; Бикельхаупт, Ф. Матиас (2021). «Происхождение асинхронности в реакциях Дильса – Альдера» . Физическая химия Химическая физика . 23 (36): 20095–20106. Бибкод : 2021PCCP...2320095V . дои : 10.1039/D1CP02456F . ПМЦ 8457343 . ПМИД 34499069 .
- ^ Хэмлин, Тревор А.; Бикельхаупт, Ф. Матиас; Фернандес, Израиль (20 апреля 2021 г.). «Концепция снижения отталкивания Паули в катализе» (PDF) . Отчеты о химических исследованиях . 54 (8): 1972–1981. doi : 10.1021/acs.accounts.1c00016 . hdl : 1871.1/a0090b38-9ab8-4c32-9d9a-b3d5de4e5ed3 . ISSN 0001-4842 . ПМИД 33759502 . S2CID 232337915 .
- ^ Вермерен, Паскаль; Бринхейс, Франсин; Хэмлин, Тревор А.; Бикельхаупт, Ф. Матиас (апрель 2020 г.). «Как щелочные катионы катализируют ароматические реакции Дильса-Альдера» . Химия: Азиатский журнал . 15 (7): 1167–1174. дои : 10.1002/asia.202000009 . ПМЦ 7187256 . ПМИД 32012430 .
- ^ Хансен, Томас; Вермерен, Паскаль; Ёсисада, Рёдзи; Филиппов Дмитрий В.; ван дер Марель, Гейсберт А.; Коди, Джерун, округ Колумбия; Хэмлин, Тревор А. (19 февраля 2021 г.). «Как кислоты Льюиса катализируют раскрытие циклооксида циклогексена» . Журнал органической химии . 86 (4): 3565–3573. дои : 10.1021/acs.joc.0c02955 . ПМК 7901664 . ПМИД 33538169 .
- ^ Тикинк, Эвелин Х.; Вермерен, Паскаль; Бикельхаупт, Ф. Матиас; Хэмлин, Тревор А. (7 октября 2021 г.). «Как кислоты Льюиса катализируют еновые реакции». Европейский журнал органической химии . 2021 (37): 5275–5283. дои : 10.1002/ejoc.202101107 . hdl : 2066/241097 . S2CID 239089361 .
- ^ Вермерен, Паскаль; Хэмлин, Тревор А.; Фернандес, Израиль; Бикельхаупт, Ф. Матиас (2020). «Происхождение повышения скорости и асинхронности в реакциях Дильса – Альдера, катализируемых иминием» . Химическая наука . 11 (31): 8105–8112. дои : 10.1039/D0SC02901G . ПМЦ 8163289 . ПМИД 34094173 .
- ^ Вермерен, Паскаль; Хэмлин, Тревор А.; Бикельхаупт, Ф. Матиас; Фернандес, Израиль (17 марта 2021 г.). «Бифункциональные реакции Дильса-Альдера, катализируемые донором водородной связи: происхождение стереоселективности и повышения скорости» . Химия: Европейский журнал . 27 (16): 5180–5190. дои : 10.1002/chem.202004496 . ПМК 8049058 . ПМИД 33169912 .
- ^ Вермерен, Паскаль; Тецца, Марко Далла; Донген, Мишель; Фернандес, Израиль; Бикельхаупт, Ф. Матиас; Хэмлин, Тревор А. (21 июля 2021 г.). «Реакции Дильса-Альдера, катализируемые кислотой Льюиса: тенденции реактивности в периодической таблице» . Химия: Европейский журнал . 27 (41): 10610–10620. дои : 10.1002/chem.202100522 . ПМК 8360170 . ПМИД 33780068 .
- ^ Уайт, Джеймс Д.; Шоу, Субрата (2011). «Цис-2,5-диаминобицикло[2.2.2]октан, новый каркас для асимметричного катализа с помощью комплексов сален-металл». Орг. Летт. 13 (9): 2488–91. дои : 10.1021/ol2007378 . ПМИД 21462988 .
- ^ Эванс, Д.А.; Чепмен, КТ; Бисаха, Дж. (1988). «Асимметричные реакции циклоприсоединения Дильса-Альдера с хиральными α,β-ненасыщенными N-ацилоксазолидинонами». Журнал Американского химического общества . 110 (4): 1238–1256. дои : 10.1021/ja00212a037 .
- ^ Кори, Э.Дж.; Ло, Т.П. (1991). «Первое применение привлекательных внутримолекулярных взаимодействий для разработки хиральных катализаторов для высокоэнантиоселективных реакций Дильса-Альдера». Журнал Американского химического общества . 113 (23): 8966–8967. дои : 10.1021/ja00023a066 .
- ^ Кори, Э.Дж.; Сибата, Т.; Ли, Т.В. (2002). «Асимметричные реакции Дильса-Альдера, катализируемые хиральным оксазаборолидином, активированным трифликовой кислотой». Журнал Американского химического общества . 124 (15): 3808–3809. дои : 10.1021/ja025848x . ПМИД 11942799 .
- ^ Рю, Д.Х.; Кори, Э.Дж. (2003). «Активация хирального оксазаборолидина трифлимидом приводит к созданию более общей каталитической системы для энантиоселективного присоединения Дильса-Альдера». Журнал Американского химического общества . 125 (21): 6388–6390. дои : 10.1021/ja035393r . ПМИД 12785777 .
- ^ Джонсон, Дж. С.; Эванс, Д.А. (2000). «Хиральные комплексы бис (оксазолина) меди (II): универсальные катализаторы для реакций энантиоселективного циклоприсоединения, реакций альдола, Михаэля и карбонильного ена». Отчеты о химических исследованиях . 33 (6): 325–335. дои : 10.1021/ar960062n . ПМИД 10891050 .
- ^ Арендт, штат Калифорния; Бортс, CJ; Макмиллан, DWC (2000). «Новые стратегии органического катализа: первая высокоэнантиоселективная органокаталитическая реакция Дильса-Альдера». Журнал Американского химического общества . 122 (17): 4243–4244. дои : 10.1021/ja000092s .
- ^ Хой, TR; Бэр, Б.; Ню, Д.; Уиллоуби, штат Пенсильвания; Вудс, БП (2012). «Гексадегидро-Реакция Дильса – Альдера» . Природа . 490 : 208–212. дои : 10.1038/nature11518 . ПМЦ 3538845 .
- ^ Флюгель, Лукас Л.; Хой, Томас Р. (2021). «Реакция гексадегидро-Дильса-Альдера: получение бензона посредством циклоизомеризации связанных триинов» . хим. Преподобный . 121 (4): 2413–2444. doi : 10.1021/acs.chemrev.0c00825 . ПМЦ 8008985 .
- ^ Минами, Ацуши; Оикава, Хидеаки (2016). «Последние достижения Дильса-Альдеразы, участвующие в биосинтезе натуральных продуктов» . Журнал антибиотиков . 69 (7): 500–506. дои : 10.1038/ja.2016.67 . ПМИД 27301662 . S2CID 30482282 .
- ^ Бер, Арно (2000). «Металлоорганические соединения и гомогенный катализ». Энциклопедия промышленной химии Ульмана . дои : 10.1002/14356007.a18_215 . ISBN 978-3527306732 .
- ^
- Дильс, О.; Олдер, К. (1928). «Синтезы в гидроароматическом ряду. I. Сообщение: Присоединения «диеновых» углеводородов». «Анналы химии» Юстуса Либиха . 460 : 98–122. дои : 10.1002/jlac.19284600106 .
- Дильс, О.; Олдер, К. (1929). «Синтезы в гидроароматическом ряду, II. Сообщение: О кантаридине». Отчеты Немецкого химического общества . 62 (3): 554–562. дои : 10.1002/cber.19290620318 .
- Дильс, О.; Олдер, К. (1929). «Синтез гидроароматического ряда, III. Связь: Синтез терпенов, камфор, гидроароматических и гетероциклических систем. Сотрудничали гг. Вольфганг Любберт, Эрих Науйокс, Франц Квербериц, Карл Рёль, Харро Зегеберг». «Анналы химии» Юстуса Либиха . 470 : 62-103. дои : 10.1002/jlac.19294700106 .
- Дильс, О.; Олдер, К. (1929). «Синтез в гидроароматическом ряду, IV. Сообщение: О добавлении малеинового ангидрида к арилированным диенам, триенам и фульвенам (под редакцией Пола Приса)». Отчеты Немецкого химического общества . 62 (8): 2081–2087. дои : 10.1002/cber.19290620829 .
- Дильс, О.; Олдер, К. (1929). «Синтезы в гидроароматическом ряду, V. О Δ4-тетрагидро-о-фталевой кислоте (заявление по сообщению Э. Х. Фармера и Ф. Л. Уоррена: Свойства сопряженных двойных связей (VII)». Отчеты Немецкого химического общества . 62 (8) . ): 2087–2090. doi : 10.1002/cber.19290620830 .
- Дильс, О.; Олдер, К. (1929). «Синтез в гидроароматическом ряду, VI. Сообщение, Курт Альдер и Герхард Штайн: О частично гидрированных нафто- и антрахинонах с водородом в положениях γ и δ. (Под редакцией Пола Приса и Ханса Винклера)». Отчеты Немецкого химического общества . 62 (8): 2337–2372. дои : 10.1002/cber.19290620872 .
- Дильс, О.; Олдер, К. (1930). «Синтез в гидроароматическом ряду, VII. Коммуникация. (Под редакцией Харрена Эрнста Петерсена и Франца Кверберица.)». «Анналы химии» Юстуса Либиха . 478 : 137–154. дои : 10.1002/jlac.19304780109 .
- Дильс, О.; Олдер, К. (1931). «Синтезы в гидроароматическом ряду, VIII. Сообщение: Диеновые синтезы антрацена. Формы антрацена». «Анналы химии» Юстуса Либиха . 486 : 191-202. дои : 10.1002/jlac.19314860110 .
- Дильс, О.; Олдер, К. (1931). «Синтезы в гидроароматическом ряду, IX. Связь: Синтез камфенилона и сантенса». «Анналы химии» Юстуса Либиха . 486 : 202-210. дои : 10.1002/jlac.19314860111 .
- Дильс, О.; Олдер, К. (1931). «Синтезы в гидроароматическом ряду, X. Связь: «Синтезы диена»︁ с пирролом и его гомологами». «Анналы химии» Юстуса Либиха . 486 : 211-225. дои : 10.1002/jlac.19314860112 .
- Дильс, О.; Олдер, К. (1931). "Синтезы в гидроароматическом ряду, XI. Сообщение. ("Диеновые синтезы" циклопентадиена, циклогексадиена и бутадиена с ацетилендикарбоновой кислотой и их эфирами". Юстус Либигс Анналы химии . 490 : 236–242. doi : 10.1002 /jlac.19314900109 .
- Дильс, О.; Олдер, К. (1931). «Синтезы в гидроароматическом ряду, XII. Связь. («Диеновые синтезы»︁ кислородсодержащих гетероциклов. 2. Диеновые синтезы фурана.)». «Анналы химии» Юстуса Либиха . 490 : 243-257. дои : 10.1002/jlac.19314900110 .
- Дильс, О.; Олдер, К. (1931). «Синтезы в гидроароматическом ряду, XIII. Связь. («Диеновые синтезы»︁ кислородсодержащих гетероциклов. 3. Диеновые синтезы кумалинов.)». «Анналы химии» Юстуса Либиха . 490 : 257-266. дои : 10.1002/jlac.19314900111 .
- Дильс, О.; Олдер, К. (1931). «Синтезы в гидроароматическом ряду, XIV. Связь. («Диеновые синтезы»︁ азотсодержащих гетероциклов. 2. Диеновые синтезы пирролов с ацетилендикарбоновыми кислотами и с их эфирами.)». «Анналы химии» Юстуса Либиха . 490 : 267-276. дои : 10.1002/jlac.19314900112 .
- Дильс, О.; Олдер, К. (1931). «Синтезы в гидроароматическом ряду, XV. Связь. («Диеновые синтезы»︁ азотсодержащих гетероциклов. 3. Диеновые синтезы индолов.)». «Анналы химии» Юстуса Либиха . 490 : 277–294. дои : 10.1002/jlac.19314900113 .
- Дильс, О.; Олдер, К. (1932). «Синтезы в гидроароматическом ряду, XVI. Сообщение. («Диеновые синтезы» азотсодержащих гетероциклов. 4. Диеновые синтезы пирролов, имидазолов и пиразолов.)». «Анналы химии» Юстуса Либиха . 498 : 1–15. дои : 10.1002/jlac.19324980102 .
- Дильс, О.; Олдер, К. (1932). «Синтезы в гидроароматическом ряду, XVII. Связь. («Диеновые синтезы» азотсодержащих гетероциклов. 5. Диеновые синтезы пиридина, хинолина, хинальдина и изохинолина.)». «Анналы химии» Юстуса Либиха . 498 : 16-49. дои : 10.1002/jlac.19324980103 .
- Дильс, О.; Олдер, К. (1933). «Синтезы гидроароматического ряда, «Анналы химии» Юстуса Либиха . 505 : 103-150. дои : 10.1002/jlac.19335050109 .
- Дильс, О.; Олдер, К. (1934). «Синтезы гидроароматического ряда, «Анналы химии» Юстуса Либиха . 510 : 87–128. дои : 10.1002/jlac.19345100106 .
- Дильс, О.; Риз, Дж. (1934). «Синтез гидроароматического ряда, XX. О присоединении эфира ацетилендикарбоновой кислоты к гидразобензолу». «Анналы химии» Юстуса Либиха . 511 : 168–182. дои : 10.1002/jlac.19345110114 .
- Дильс, О.; Мейер, Р. (1934). «Синтезы в гидроароматическом ряду XXI. «Диеновые синтезы» азотсодержащих гетероциклов. 8. О ходе диенового синтеза пиридина в растворе метилового спирта». «Анналы химии» Юстуса Либиха . 513 : 129–145. дои : 10.1002/jlac.19345130108 .
- Дильс, О.; Фридрихсен, В. (1934). «Синтез гидроароматического ряда XXII. Об аддуктах антрацен-C4O3, их пригодности для диенового синтеза и новом принципе синтеза фталевых кислот и дигидрофталевых кислот». «Анналы химии» Юстуса Либиха . 513 : 145–155. дои : 10.1002/jlac.19345130109 .
- Дильс, О.; Мёллер, Ф. (1935). «Синтезы гидроароматического ряда, XXIII. «Диеновые синтезы» азотсодержащих гетероциклов. 9. Стильбазол и ацетилендикарбоновые эфиры». «Анналы химии» Юстуса Либиха . 516 : 45–61. дои : 10.1002/jlac.19355160104 .
- Дильс, О.; Кех, Х. (1935). «Синтезы гидроароматического ряда, XXIV «Диеновые синтезы» азотсодержащих гетероциклов». «Анналы химии» Юстуса Либиха . 519 : 140–146. дои : 10.1002/jlac.19355190112 .
- Дильс, О.; Риз, Дж. (1935). «Синтез гидроароматического ряда, XXV Об аддуктах эфира ацетилендикарбоновой кислоты и гидразосоединений (2)». «Анналы химии» Юстуса Либиха . 519 : 147–157. дои : 10.1002/jlac.19355190113 .
- Дильс, О.; Хармс, Дж. (1935). «Синтезы гидроароматического ряда, XXVI. «Диеновые синтезы» азотсодержащих гетероциклов. 11. Об аддуктах, образующихся из изохинолина и эфира ацетилендикарбоновой кислоты». «Анналы химии» Юстуса Либиха . 525 :73–94. дои : 10.1002/jlac.19365250107 .
- Дильс, О.; Шрум, Х. (1937). «Синтезы в гидроароматическом ряду, XXVII. «Диеновые синтезы» азотсодержащих гетероциклов. 12. О деградации «желтого вещества»︁ до изомера норлупинана (1-метил-октагидро-индолизин)». «Анналы химии» Юстуса Либиха . 530 : 68–86. дои : 10.1002/jlac.19375300106 .
- Дильс, О.; Пистор, Х. (1937). «Синтезы гидроароматического ряда, XXVIII. «Диеновые синтезы» азотсодержащих гетероциклов. 13. Эфиры α-пиколина и ацетилендикарбоновых кислот». «Анналы химии» Юстуса Либиха . 530 :87–98. дои : 10.1002/jlac.19375300107 .
- ^ «Нобелевская премия по химии 1950 года» . Нобелевский фонд . Проверено 19 февраля 2016 г.
- ^ Вудворд, РБ; Сондхаймер, Ф.; Тауб, Д.; Хойслер, К.; Макламор, WM (1952). «Полный синтез стероидов». Журнал Американского химического общества . 74 (17): 4223–4251. дои : 10.1021/ja01137a001 .
- ^ Кори, Э.Дж.; Вайншенкер, Нью-Мексико; Шааф, ТК; Хубер, В. (1969). «Стереоконтролируемый синтез простагландинов F-2a и E-2 (dl)». Журнал Американского химического общества . 91 (20): 5675–7. дои : 10.1021/ja01048a062 . ПМИД 5808505 .
- ^ Данишевский, С.; Хирама, М.; Фрич, Н.; Кларди, Дж. (1979). «Синтез префената динатрия и эпипрефената динатрия. Стереохимия префеновой кислоты и наблюдение катализируемой основаниями перегруппировки префеновой кислоты в п-гидроксифенилмолочную кислоту». Журнал Американского химического общества . 101 (23): 7013–7018. дои : 10.1021/ja00517a039 .
- ^ Вендер, Пенсильвания; Шаус, Дж. М.; Уайт, AW (1980). «Общая методология синтеза цис-гидроизохинолина: Синтез резерпина». Журнал Американского химического общества . 102 (19): 6157–6159. дои : 10.1021/ja00539a038 .
- ^ Мартин, Сан-Франциско; Рюгер, Х.; Уильямсон, ЮАР; Гжейщак, С. (1987). «Общие стратегии синтеза индольных алкалоидов. Полный синтез (±)-резерпина и (±)-α-йохимбина». Журнал Американского химического общества . 109 (20): 6124–6134. дои : 10.1021/ja00254a036 .
- ^ Николау, КЦ; Ян, З.; Лю, Джей-Джей; Уэно, Х.; Нантермет, П.Г.; Гай, РК; Клэйборн, CF; Рено, Дж.; Куладурос, Э.А.; Полваннан, К.; Соренсен, Э.Дж. (1994). «Тотальный синтез таксола». Природа . 367 (6464): 630–4. Бибкод : 1994Natur.367..630N . дои : 10.1038/367630a0 . ПМИД 7906395 . S2CID 4371975 .
- ^ Нарасака, К.; Шимада, С.; Осода, К.; Ивасава, Н. (1991). «Фенилбороновая кислота как шаблон в реакции Дильса-Альдера». Синтез . 1991 (12): 1171–1172. дои : 10.1055/s-1991-28413 .
- ^ Смит, AB; Сестело, JP; Дормер, П.Г. (1995). «Тотальный синтез (-)-фурахиноцина С». Журнал Американского химического общества . 117 (43): 10755–10756. дои : 10.1021/ja00148a023 .
- ^ Козьмин, С.А.; Равал, В.Х. (1998). «Общая стратегия в отношении алкалоидов аспидоспермы: эффективный стереоконтролируемый синтез таберсонина». Журнал Американского химического общества . 120 (51): 13523–13524. дои : 10.1021/ja983198k .
- ^ Гиббс, РА; Окамура, WH (1988). «Краткий энантиоселективный синтез (+)-стерпурена: полный внутримолекулярный перенос центральных к аксиальным и центральным хиральным элементам». Журнал Американского химического общества . 110 (12): 4062–4063. дои : 10.1021/ja00220a069 .
- ^ Чарест, МГ; Сигел, доктор медицинских наук; Майерс, АГ (2005). «Синтез (-)-тетрациклина». Журнал Американского химического общества . 127 (23): 8292–3. дои : 10.1021/ja052151d . ПМИД 15941256 .
- ^ Даубен, В.Г.; Кессель, ЧР; Такемура, К.Х. (1980). «Простой и эффективный полный синтез кантаридина с помощью реакции Дильса-Альдера под высоким давлением». Журнал Американского химического общества . 102 (22): 6893–6894. дои : 10.1021/ja00542a060 .
- ^ Холмс, HL (1948). «Реакция Дильса-Альдера, этиленовые и ацетиленовые диенофилы». Органические реакции . Том. 4. С. 60–173. дои : 10.1002/0471264180.или004.02 . ISBN 978-0471264187 .
- ^ Бутц, Л.В.; Рытина, А. В. (1949). «Хиноны реакции Дильса-Альдера и другие цикленоны». Органические реакции . Том. 5. С. 136–192. дои : 10.1002/0471264180.или005.03 . ISBN 978-0471264187 .
- ^ Клёцель, MC (1948). «Реакция Дильса-Альдера с малеиновым ангидридом». Органические реакции . Том. 4. стр. 1–59. дои : 10.1002/0471264180.или004.01 . ISBN 978-0471264187 .
- ^ Хайнцельман, Греция; Мей, ИК; Махаджан, Ю.Р.; Вайнреб, SW (2005). «Реакции Дильса-Альдера иминодиенофилов». Органические реакции . Том. 65. стр. 141–599. дои : 10.1002/0471264180.или065.02 . ISBN 978-0471264187 .
- ^ Цыганек, Э. (1984). «Внутримолекулярная реакция Дильса-Альдера». Органические реакции . Том. 32. С. 1–374. дои : 10.1002/0471264180.или032.01 . ISBN 978-0471264187 .
Библиография
[ редактировать ]- Кэри, Фрэнсис А.; Сундберг, Ричард Дж. (2007). Продвинутая органическая химия: Часть B: Реакции и синтез (5-е изд.). Нью-Йорк: Спрингер. ISBN 978-0387683546 .
Внешние ссылки
[ редактировать ]- [1] Английский перевод основополагающей немецкой статьи Дильса и Альдера 1928 года, которая принесла им Нобелевскую премию. Английское название: «Синтез гидроароматического ряда»; Немецкое название «Synthesen in der Hydroaromatischen Reihe».
| Базы данных органов управления : Национальные |
|---|