Теория решений Флори – Хаггинса
| Полимерная наука |
|---|
 |

Теория растворов Флори-Хаггинса представляет собой решетчатую модель термодинамики которая , растворов полимеров учитывает большое различие размеров молекул при адаптации обычного выражения для энтропии смешения . Результатом является уравнение свободной энергии Гиббса. изменения для смешивания полимера с растворителем . Хотя он делает упрощающие предположения, он дает полезные результаты для интерпретации экспериментов.

Теория [ править ]
Термодинамическое уравнение при изменения энергии Гиббса смешивании при постоянной температуре и (внешнем) давлении имеет вид

Изменение, обозначаемое , — значение переменной . для раствора или смеси за вычетом значений для чистых компонентов, рассматриваемых отдельно Цель состоит в том, чтобы найти явные формулы для и , приращения энтальпии и энтропии смешивания , связанные с процессом .



Результат, полученный Флори [1] и Хаггинс [2] является
![{\displaystyle \Delta G_{\rm {mix}}=RT[\,n_{1}\ln \phi _{1}+n_{2}\ln \phi _{2}+n_{1}\phi _{2}\кто _{12}\,]\,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/043a3377fef83a6b6ec3f97120538f2aaafec284)
Правая часть является функцией количества родинок. и объемная доля растворителя ( компонент ), количество молей и объемная доля полимера (компонент ), с введением параметра учитывать энергию взаимодиспергирующих молекул полимера и растворителя. газовая постоянная и это абсолютная температура . Объемная доля аналогична мольной доле , но взвешена с учетом относительных размеров молекул. Для небольшого растворенного вещества вместо этого появятся мольные доли, и эта модификация является новшеством Флори и Хаггинса. В наиболее общем случае параметр смешивания , является параметром свободной энергии, таким образом, включая энтропийную составляющую. [1] [2]









Вывод [ править ]
Сначала мы вычисляем энтропию смешивания , увеличение неопределенности относительно местоположения молекул при их рассредоточении. В чистых конденсированных фазах — растворителе и полимере — куда бы мы ни посмотрели, мы находим молекулу. [3] Конечно, любая идея «найти» молекулу в заданном месте — это мысленный эксперимент , поскольку на самом деле мы не можем исследовать пространственные местоположения размером с молекулу. Выражение энтропии смешения растворенное малых молекул в мольных долях больше не является разумным, когда вещество представляет собой макромолекулярную цепь . Мы учитываем эту диссимметрию размеров молекул, предполагая, что отдельные сегменты полимера и отдельные молекулы растворителя занимают узлы в решетке . Каждый узел занят ровно одной молекулой растворителя или одним мономером полимерной цепи, поэтому общее число узлов равно

- число молекул растворителя и – количество молекул полимера, каждая из которых имеет сегменты. [4]



Для случайного блуждания по решетке [3] мы можем рассчитать изменение энтропии (увеличение пространственной неопределенности ) в результате смешивания растворенного вещества и растворителя.
![{\displaystyle \Delta S_{\rm {mix}}=-k_{\rm {B}}\left[N_{1}\ln {\tfrac {N_{1}}{N}}+N_{2} \ln {\tfrac {xN_{2}}{N}}\right]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1f4a5b0bcda7d8f77d6a52cbfbcee2ce149b9586)
где – постоянная Больцмана . решетки Определите объемные доли и


Это также вероятности того, что данный узел решетки, выбранный случайно , занят молекулой растворителя или сегментом полимера соответственно. Таким образом
![{\displaystyle \Delta S_{\rm {mix}}=-k_{\rm {B}}[\,N_{1}\ln \phi _{1}+N_{2}\ln \phi _{2 }\,]\,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b9a58a81330833f0cec1b7cc8cf68ab86476dce6)
Для небольшого растворенного вещества, молекулы которого занимают всего один узел решетки, равна единице, объемные доли уменьшаются до молекулярных или мольных долей , и мы восстанавливаем обычную энтропию смешения .
Помимо энтропийного эффекта, мы можем ожидать изменения энтальпии . [5] Следует учитывать три молекулярных взаимодействия: растворитель-растворитель. , мономер-мономер (не ковалентная связь , а между разными участками цепи), и мономер-растворитель . Каждый из последних происходит за счет среднего значения двух других, поэтому приращение энергии на контакт мономер-растворитель составляет




Общее количество таких контактов

где — координационное число, число ближайших соседей узла решетки, каждый из которых занят либо одним сегментом цепи, либо молекулой растворителя. То есть, – общее количество полимерных сегментов (мономеров) в растворе, поэтому – число ближайших соседей ко всем сегментам полимера. Умножение на вероятность что любой такой сайт занят молекулой растворителя, [6] получаем общее число молекулярных взаимодействий полимер-растворитель. аппроксимация в соответствии с теорией среднего поля Следуя этой процедуре, делается , тем самым сводя сложную проблему многих взаимодействий к более простой проблеме одного взаимодействия.



Изменение энтальпии равно изменению энергии за взаимодействие мономера полимера с растворителем, умноженному на количество таких взаимодействий.

Параметр взаимодействия полимер-растворитель chi определяется как

Он зависит от природы растворителя и растворенного вещества и является единственным зависящим от материала параметром модели, . Изменение энтальпии становится

С точки зрения ассемблера, полное изменение свободной энергии равно
где мы преобразовали выражение из молекул и к кротам и путем переноса постоянной Авогадро к газовой постоянной .


Значение параметра взаимодействия можно оценить по параметрам растворимости Хильдебранда и



где – фактический объем полимерного сегмента.

В самом общем случае взаимодействие и полученный параметр смешивания, , является параметром свободной энергии, таким образом, включая энтропийную составляющую. [1] [2] Это означает, что помимо обычной энтропии смешения существует еще один энтропийный вклад от взаимодействия растворителя и мономера. Этот вклад иногда очень важен для количественного предсказания термодинамических свойств.

Существуют более продвинутые теории решения, такие как теория Флори – Кригбаума .
Разделение фаз жидкость-жидкость [ править ]
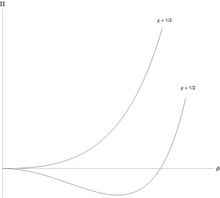
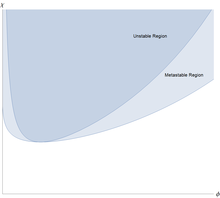
Полимеры могут отделяться от растворителя, и делают это характерным образом. [4] Свободная энергия Флори – Хаггинса на единицу объема для полимера с мономеры, можно записать в простой безразмерной форме


для объемная доля мономеров и . Осмотическое давление (в приведенных единицах) равно


- .

Раствор полимера устойчив по отношению к небольшим колебаниям, когда вторая производная этой свободной энергии положительна. Эта вторая производная

и решение сначала становится неустойчивым, когда эта и третья производная

оба равны нулю. Затем немного алгебры показывает, что раствор полимера сначала становится нестабильным в критической точке при

Это означает, что для всех значений эффективное взаимодействие мономер-растворитель имеет слабое отталкивание, но оно слишком слабое, чтобы вызвать разделение жидкость/жидкость. Однако, когда происходит разделение на две сосуществующие фазы, одна из которых богаче полимером, но беднее растворителем, чем другая.


Необычной особенностью разделения фаз жидкость/жидкость является его сильная асимметричность: объемная доля мономеров в критической точке примерно равна , что очень мало для крупных полимеров. Количество полимера в сосуществующей фазе с высоким содержанием растворителя и низким содержанием полимера чрезвычайно мало для длинных полимеров. Фаза, обогащенная растворителем, близка к чистому растворителю. Это свойственно полимерам: смесь малых молекул можно аппроксимировать выражением Флори – Хаггинса с помощью , а потом и обе сосуществующие фазы далеки от чистоты.



Полимерные смеси [ править ]
Синтетические полимеры редко состоят из цепей одинаковой длины в растворителе. Плотность свободной энергии Флори-Хаггинса можно обобщить. [5] к N-компонентной смеси полимеров длиной к


Для бинарной полимерной смеси , где один вид состоит из мономеры и другие мономеры это упрощает до


Как и в случае с разбавленными растворами полимеров, первые два члена в правой части представляют собой энтропию смешения. Для крупных полимеров и эти члены пренебрежимо малы. Это означает, что для существования устойчивой смеси , поэтому для смешивания полимеров A и B их сегменты должны притягиваться друг к другу. [6]



Ограничения [ править ]
Теория Флори-Хаггинса имеет тенденцию хорошо согласовываться с экспериментами в режиме полуразбавленной концентрации и может использоваться для подбора данных для еще более сложных смесей с более высокими концентрациями. Теория качественно предсказывает фазовое разделение, тенденцию к несмешиванию высокомолекулярных частиц, зависимость взаимодействия от температуры и другие особенности, обычно наблюдаемые в полимерных смесях. Однако немодифицированная теория Флори-Хаггинса не может предсказать более низкую критическую температуру растворения, наблюдаемую в некоторых полимерных смесях, и отсутствие зависимости критической температуры. по длине цепи . [7] Кроме того, можно показать, что для бинарной смеси полимеров с одинаковой длиной цепи критическая концентрация должна быть ; однако наблюдались смеси полимеров, в которых этот параметр сильно асимметричен. В некоторых смесях энтропия смешения может доминировать над взаимодействием мономеров. Приняв приближение среднего поля, Сложная зависимость параметров от температуры , состава смеси и длины цепи была исключена. В частности, взаимодействия за пределами ближайшего соседа могут иметь большое значение для поведения смеси, а распределение сегментов полимера не обязательно является однородным, поэтому в определенных узлах решетки могут возникать энергии взаимодействия, отличные от тех, которые аппроксимируются теорией среднего поля.




Один хорошо изученный [4] [6] Эффектом на энергии взаимодействия, которым пренебрегает немодифицированная теория Флори – Хаггинса, является цепная корреляция. В разбавленных полимерных смесях, где цепи хорошо разделены, внутримолекулярные силы между мономерами полимерной цепи доминируют и вызывают расслоение, приводящее к образованию областей с высокой концентрацией полимера. По мере увеличения концентрации полимера цепи имеют тенденцию перекрываться, и эффект становится менее значимым. Фактически, граница между разбавленными и полуразбавленными растворами обычно определяется концентрацией, при которой полимеры начинают перекрываться. который можно оценить как


Здесь m — масса одиночной полимерной цепи, а цепи — радиус вращения .

Ссылки [ править ]
- ^ Jump up to: Перейти обратно: а б Берчард, В. (1983). «Термодинамика раствора неионных водорастворимых полимеров». В Финч, К. (ред.). Химия и технология водорастворимых полимеров . Спрингер. стр. 125–142. ISBN 978-1-4757-9661-2 .
- ^ Jump up to: Перейти обратно: а б Фрэнкс, Ф. (1983). «Растворимость в воде и чувствительность – эффекты гидратации». В Финч, К. (ред.). Химия и технология водорастворимых полимеров . Спрингер. стр. 157–178. ISBN 978-1-4757-9661-2 .
- ^ Дейк, Менно А. ван; Пробудитесь, Андре (14 января 1998 г.). Концепции термодинамики полимеров . ЦРК Пресс. стр. 61–65. ISBN 978-1-56676-623-4 .
- ^ Jump up to: Перейти обратно: а б де Женн, Пьер-Жиль (1979). Концепции масштабирования в физике полимеров . Итака, Нью-Йорк: Издательство Корнельского университета. ISBN 080141203X . OCLC 4494721 .
- ^ Берри, Дж; и др. (2018). «Физические принципы внутриклеточной организации посредством активных и пассивных фазовых переходов». Отчеты о прогрессе в физике . 81 (46601): 046601. Бибкод : 2018RPPh...81d6601B . дои : 10.1088/1361-6633/aaa61e . ПМИД 29313527 . S2CID 4039711 .
- ^ Jump up to: Перейти обратно: а б Дой, Масао (2013). Физика мягких материалов . Грейт-Кларендон-стрит, Оксфорд, Великобритания: Издательство Оксфордского университета. ISBN 9780199652952 .
- ^ Шмид, Фридерика (2010). «Теория и моделирование многофазных полимерных систем». arXiv : 1001.1265 [ cond-mat.soft ].
Внешние ссылки [ править ]
- «Конформации, растворы и молекулярная масса» (глава книги) , глава 3 названия книги: Наука и технология полимеров; Джоэл Р. Фрид; 2-е издание, 2003 г.
Сноски [ править ]
- ^ « Термодинамика высоких полимеров растворов », Пола Дж. Флори Журнал химической физики , август 1941 г., том 9, выпуск 8, стр. 660 Аннотация . Флори предположил, что имя Хаггинса должно быть первым, поскольку он опубликовал несколько месяцев назад: Флори, П.Дж., «Термодинамика растворов высоких полимеров», J. Chem. Физ. 10 :51-61 (1942) Citation Classic № 18, 6 мая 1985 г.
- ^ «Растворы соединений с длинной цепью », Мориса Л. Хаггинса Журнал химической физики , май 1941 г., том 9, выпуск 5, с. 440 Аннотация
- ^ Мы игнорируем свободный объем из-за молекулярного беспорядка в жидкостях и аморфных твердых телах по сравнению с кристаллами . Это, а также предположение о том, что мономеры и молекулы растворенного вещества на самом деле имеют одинаковый размер, являются основными геометрическими приближениями в этой модели.
- ^ Для настоящего синтетического полимера существует статистическое распределение , длин цепей поэтому было бы среднее значение .
- ^ Энтальпия , - это внутренняя энергия скорректированная на любую давление - объем работу при постоянной (внешней) . Мы не делаем здесь никаких различий. Это позволяет аппроксимировать свободную энергию Гельмгольца , которая является естественной формой свободной энергии из теории решетки Флори-Хаггинса, к свободной энергии Гиббса.
- ^ Фактически, два участка, прилегающие к сегменту полимера, заняты другими сегментами полимера, поскольку он является частью цепи ; и еще один, то есть три, для ветвления сайтов , но только один для терминалов .
