Губчатая дегенерация центральной нервной системы
| Губчатая дегенерация центральной нервной системы | |
|---|---|
| Другие имена | Болезнь Канавана, тип Ван Богерта-Бертрана, дефицит аспартоацилазы |
 | |
| Магнитно-резонансная томография показывает дисмиелинизацию, возможный индикатор болезни Канавана. | |
| Специальность | Неврология |
| Симптомы | Гипотония, макроцефалия, потеря зрения, нарушения двигательных рефлексов, затруднения дыхания и глотания. |
| Обычное начало | 3-6 месяцев |
| Продолжительность | Терминал |
| Типы | Инфантильный, врожденный, юношеский |
| Причины | Генетический (аутосомно-рецессивный) |
| Факторы риска | Семейный анамнез (генетика) |
| Метод диагностики | Нейровизуализация, исследование мочи |
| Профилактика | Анализ ДНК, пренатальный анализ |
| Уход | Только паллиативные меры |
| Медикамент | Н/Д |
Губчатая дегенерация центральной нервной системы , также известная как болезнь Канавана , тип Ван Богерта-Бертрана или дефицит аспартоацилазы (AspA) , является редким аутосомно-рецессивным нейродегенеративным заболеванием . [1] Он принадлежит к группе генетических заболеваний, известных как лейкодистрофии . [1] где рост и поддержание миелиновой оболочки в центральной нервной системе (ЦНС) нарушены. [2] Существует три типа губчатой дегенерации: инфантильная, врожденная и ювенильная, причем ювенильная является наиболее тяжелым типом. [3] Общие симптомы у младенцев включают отсутствие двигательных навыков , слабый мышечный тонус и макроцефалию . [4] Это также может сопровождаться трудностями при кормлении и глотании, судорогами и нарушениями сна . [4] Заболевшие дети обычно умирают в возрасте до 10 лет, но продолжительность жизни может варьироваться. [5]
Причиной губчатой дегенерации ЦНС является мутация гена, кодирующего аспартоацилазу (AspA) — фермент , гидролизующий ) N-ацетиласпарагиновую кислоту (NAA . [6] В отсутствие AspA NAA накапливается и приводит к губчатой дегенерации. [7] Точные патофизиологические причины заболевания в настоящее время неясны, но существуют развивающиеся теории. [8] Губчатую дегенерацию можно диагностировать с помощью методов нейровизуализации и исследования мочи . [9] В настоящее время не существует лечения губчатой дегенерации, но исследования по использованию генной терапии для лечения этого заболевания продолжаются. [8] Губчатая дегенерация более распространена среди евреев-ашкенази , частота встречаемости среди этой этнической группы составляет 1/6000. [10]
Клинические симптомы
[ редактировать ]Губчатая дегенерация ЦНС подразделяется на три типа: инфантильную, ювенильную и врожденную; в зависимости от возраста начала заболевания и тяжести симптомов.
Инфантильный тип
[ редактировать ]Инфантильный тип — наиболее распространенный тип губчатой дегенерации ЦНС. [11] Обычно пораженные младенцы выглядят нормальными в течение первых нескольких месяцев жизни. [12] Возраст начала заболевания составляет около 6 месяцев, когда у младенцев начинают развиваться заметные психомоторные дефекты . [12] Затрагиваются различные двигательные навыки, такие как переворачивание и стабилизация движений головы. [11] гипотония и макроцефалия. В первые несколько месяцев наблюдаются также [13]
Во второй половине первого года жизни глаза большинства детей не реагируют на зрительные раздражители , наблюдаются эпизодические саккадические движения глаз , что приводит к слепоте большинства детей на втором году жизни. [5]
Симптомами терминальной стадии развития заболевания являются потливость, рвота , гипертермия , судороги и артериальная гипотония , что обычно приводит к смерти ребенка. [13] Продолжительность жизни пострадавших младенцев варьируется, но большинство младенцев не доживают до десяти лет. [5]

Врожденный тип
[ редактировать ]Возраст начала заболевания обычно составляет несколько дней после рождения при врожденном типе. Беременность и роды не затрагиваются, и ребенок рождается с нормальной внешностью и без проблем со здоровьем. [12] Однако в последующие дни пострадавшие младенцы могут стать вялыми и испытывать трудности с такими движениями, как сосание и глотание. [14] По мере прогрессирования заболевания у пациентов может наблюдаться снижение мышечного тонуса и инактивация рефлекса Моро , также известного как рефлекс испуга. [12] Это может привести к развитию дыхания Чейн-Стокса через несколько недель или даже дней после родов, что может привести к летальному исходу. [12]
Ювенильный тип
[ редактировать ]Возраст начала ювенильного типа – около пяти лет. Большинство пациентов с ювенильным типом доживают до позднего подросткового возраста . [15] У пораженных малышей обычно развивается прогрессирующий мозжечковый синдром и ухудшение умственного развития, за которым следует потеря зрения, атрофия зрительных нервов и генерализованная спастичность . [16] В отличие от инфантильной формы, макроцефалия не проявляется. [12]
Патофизиология
[ редактировать ]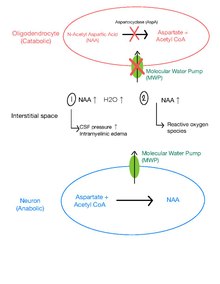
Хотя патофизиологические причины симптомов БК до сих пор неясны, развиваются теории о причинах проблем миелинизации, студенистого кортикального белого вещества и судорог. [8]
Проблемы с миелинизацией
[ редактировать ]Молекулярный водяной насос (МВП) и дисбаланс осмолитов
[ редактировать ]Повышенное давление спинномозговой жидкости (СМЖ) и внутримиелиновый отек у пациентов с БК позволяют предположить существование эффективного МВП в головном мозге. [17] [8] MWP представляет собой мембранный белок , ответственный за перекачку молекул воды вместе с растворенными молекулами NAA из внутринейронного пространства в интерстициальное пространство . [10] У здоровых людей NAA сначала транспортируется вниз по градиенту концентрации через MWP от нейронов в интерстициальное пространство и затем гидролизуется AspA в соседних олигодендроцитах . [10]
Предполагается, что у пациентов с БК дефицит AspA вызывает накопление NAA в интерстициальном пространстве, вызывая осмолитный дисбаланс и накопление воды в интерстициальном пространстве. [8] Это увеличивает гидростатическое давление между межламеллярными пространствами и внеклеточным периаксональным и паренхиматозным пространством, ослабляя плотные соединения между ними и вызывая тем самым внутримиелиновый отек. [18] Последующая демиелинизация , возможно, способствует образованию свободных пространств в белом веществе или губчатой дегенерации. [8]
Дисмиелинизация
[ редактировать ]полученные из NAA, Ацетаты, участвуют в синтезе жирных кислот , которые впоследствии включаются в липиды миелина . [3] [8] Предполагается, что у пациентов с БК дефицит AspA снижает образование ацетатов NAA и, следовательно, снижает синтез миелин-ассоциированных липидов. [6] Это приводит к дисмиелинизации , что способствует образованию вакуолей в интерстициальном пространстве и губчатой дистрофии. [8] Однако было показано, что губчатая дегенерация не вызвана напрямую нарушением синтеза миелина. [19] Модели на животных показывают, что миелинизация все еще может происходить у AspA, лишенных видов, возможно, из-за параллельных путей миелинизации на начальных стадиях миелиногенеза . [19]
Сворачивание и стабилизация белка
[ редактировать ]Дефицит AspA снижает экспрессию ацетил-коэнзима А (КоА) в клетках, который может отвечать за стабилизацию и правильное сворачивание белков . [20] Это приводит к деградации белка, особенно сильному эффекту в олигодендроцитах. [20] В исследованиях на животных видов с дефицитом AspA было показано, что деградация белка в олигодендроцитах вызывает серьезную потерю белков миелина. [21]
Желатиновое подкорковое белое вещество
[ редактировать ]Дефицит AspA, который жизненно важен в олигодендроцитах для производства ацетата, полученного из NAA, приводит к отсутствию регуляции генетической структуры и экспрессии в этих клетках. [1] Это приводит к гибели олигодендроцитов, следовательно, вызывает повреждение нейронов и образование вакуолей в подкорковом веществе . [22] Эти вакуоли способствуют образованию подкоркового белого вещества студенистой текстуры, обнаруженного у многих пациентов с БК. [22]
Судороги и нейродегенерация
[ редактировать ]Патофизиологические причины судорог и нейродегенерации у пациентов с БК, вероятно, связаны с окислительным стрессом, вызванным накоплением NAA. [23] Предполагается, что NAA способствует окислительному стрессу за счет стимулирования активных форм кислорода , а также снижения неферментативной антиоксидантной защиты. [23] NAA также влияет на несколько антиоксидантных ферментов, таких как каталаза и глутатионпероксидаза , ухудшая детоксикацию перекиси водорода . [24] Недавние исследования на животных показали, что хронический окислительный стресс может вызывать дисфункцию митохондрий , делая мозг более восприимчивым к эпилептическим припадкам . [24] [25]
Диагностика
[ редактировать ]Болезнь Канавана первоначально распознается по появлению симптомов, однако для окончательного диагноза необходимы дальнейшие обследования. Методы нейровизуализации, такие как компьютерная томография (КТ) или магнитно-резонансная томография (МРТ), обычно используются для обнаружения наличия дегенеративного подкоркового белого вещества . [26] Для диагностики также может быть использована микроскопия спинномозговой жидкости, при которой набухшие астроциты с деформированными и удлиненными митохондриями. у пациентов можно увидеть [5]
Исследования мочи используются для дифференциации пациентов с БК от других нейродегенеративных заболеваний со схожей морфологией , таких как болезнь Александера и болезнь Тея-Сакса (которые аналогичным образом демонстрируют макроцефалию ), поскольку у пациентов с БК однозначно наблюдается повышенная экскреция NAA. [5] [13]
Профилактика
[ редактировать ]
Анализ ДНК обычно используется для определения того, являются ли родители носителями мутантного гена. [27] Пренатальная диагностика с помощью анализа ДНК или определения NAA в околоплодных водах (количество которых может увеличиваться при нарушенной беременности) также может использоваться, когда анализ ДНК не может быть проведен у родителей. [28] Было замечено, что среди еврейского населения ашкенази наблюдается аномально высокий уровень носителей. [4] Риск губчатой дегенерации у их потомства составляет один из четырех, если оба родителя являются носителями мутантного гена. [28]
Методы лечения
[ редактировать ]В настоящее время не известны конкретные формы лечения губчатой дегенерации ЦНС. [29] Некоторые модули лечения проходят экспериментальные испытания, а нынешним пациентам оказывают паллиативную помощь, все из которых описаны ниже.
Текущие паллиативные меры
[ редактировать ]Текущие пациенты поддерживаются рекомендациями по лечению других педиатрических нейродегенеративных заболеваний. [30] У пациентов с респираторными заболеваниями аспираторы . используются для удаления слизи из верхних дыхательных путей легких [8] Концентраторы кислорода также вводятся для очистки дыхательных путей и непрерывной подачи воздуха для облегчения дыхания. [8] Что касается младенцев с гипотонией, ее решают путем предоставления оборудования для позиционирования, такого как специализированные коляски, кресла для купания и сиденья для кормления. [31]
Возможные модули лечения в стадии разработки
[ редактировать ]Внутрибрюшинные инъекции липоевой кислоты
[ редактировать ]Липоевая кислота (которая может проникать через гематоэнцефалический барьер) недавно была опробована в доклинических исследованиях , в ходе которых ее вводили треморным крысам внутрибрюшинно . [32] Крысы с тремором считаются естественной моделью губчатой дегенерации ЦНС, поскольку NAA вызывает окислительный стресс. [33] В результате этих исследований были получены положительные результаты, позволяющие предположить, что липоевая кислота может быть возможным подходом для симптоматического лечения . [32]
Внутрибрюшинное введение лития
[ редактировать ]Возможным лечением является использование нейропротекторных методов для компенсации неврологического повреждения ЦНС, вызванного накоплением NAA. [8] Одним из потенциальных методов лечения, которое было выявлено, является литий , который, как было замечено, оказывает нейропротекторное действие у пациентов с деменцией . [34] Внутрибрюшинное введение лития было протестировано как на крысах с тремором, так и на крысах дикого типа, что привело к снижению уровней NAA у обоих видов. [35] В ходе испытаний на людях было обнаружено, что уровни NAA в мозге и моче пациента снижаются после года лечения. [29] Это сочетается с повышением бдительности и визуальным отслеживанием. [29] Однако. Симптомы БК, включая аксиальную гипотонию и спастическую диплегию, сохранялись. [8]

Генная терапия
[ редактировать ]Поскольку БК возникает в результате моногенного дефекта и локализуется в ЦНС, потенциальным методом лечения является заместительная генная терапия. [8] Эта терапия включает замену мутантного гена заболевания полностью функциональным геном с использованием вектора, который транспортирует терапевтическую ДНК в клетки, позволяя клеткам продуцировать AspA. [36] Аденоассоциированные вирусы (ААВ) широко используются в качестве векторов для генной терапии. [8] Их взяли на вооружение, поскольку они не воспроизводят себя и почти нетоксичны. [8] два серотипа Для лечения используются : AAV2 и AAV9. [37] Разница стереотипов в том, что AAV2 ограничен гематоэнцефалическим барьером (ГЭБ), тогда как AAV9 может пересекать ГЭБ, что позволяет проводить лечение даже на поздних стадиях заболевания. [38] Однако текущие исследования показывают, что AAV могут вызывать нежелательные иммунные реакции у младенцев и имеют ограниченную способность инкапсулировать гены. [39]
Эпидемиология
[ редактировать ]Губчатая дегенерация ЦНС носит панэтнический характер , что обусловлено ее распространенностью среди евреев-ашкенази . Среди них обнаружены две распространенные мутации: миссенс-мутация (Glu285AIa) и нонсенс-мутация (Tyr231X). [40] При миссенс-мутации происходит замена глутаминовой кислоты на аланин . [41] Что касается нонсенс-мутации, то тирозиновый кодон заменяется терминирующим кодоном . [41] Генетический скрининг показывает, что примерно 1 из 40 здоровых евреев является носителем, а заболеваемость этой болезнью в этой группе населения достигает 1 из 6000. [10]
История
[ редактировать ]О первом случае губчатой дегенерации ЦНС сообщили в 1928 году Глобус и Штраус. [42] который назвал этот случай болезнью Шильдера, термином для диффузного миелинокластического склероза . [43] [44] [45] В 1931 году Канаван сообщил о случае, когда мегалэнцефалия при дегенерации головного мозга отличается от мегалэнцефалии, вызванной опухолью . [46] Однако ей не удалось распознать губчатые изменения, которые указывают на уникальную патологическую причину, отличающую ее случай от болезни Шильдера. [47] Позже, в 1937 году, Эйслебергль сообщила о шести случаях в еврейских семьях и обнаружила семейные характеристики губчатой дегенерации, но классифицировала эти случаи как склероз Краббе . [47] [48] Лишь в 1949 году Ван Богерт и Бертран сообщили о пяти случаях в еврейских семьях, после чего дальнейший патологический анализ подтвердил, что губчатая дегенерация является нозологической единицей. [47]
Ссылки
[ редактировать ]- ^ Перейти обратно: а б с Баслоу М.Х., Гилфойл Д.Н. (апрель 2013 г.). «Болезнь Канавана, редкая губчатая лейкодистрофия человека с ранним началом: понимание ее происхождения и возможных клинических вмешательств». Биохимия . 95 (4): 946–56. дои : 10.1016/j.biochi.2012.10.023 . ПМИД 23151389 .
- ^ «Болезнь Канавана» . НОРД (Национальная организация по редким заболеваниям) . Проверено 31 марта 2021 г.
- ^ Перейти обратно: а б Намбудири А.М., Питхамбаран А., Мэтью Р., Самбху П.А., Хершфилд Дж., Моффетт Дж.Р., Мадхаварао К.Н. (июнь 2006 г.). «Болезнь Канавана и роль N-ацетиласпартата в синтезе миелина». Молекулярная и клеточная эндокринология . 252 (1–2): 216–23. дои : 10.1016/j.mce.2006.03.016 . ПМИД 16647192 . S2CID 12255670 .
- ^ Перейти обратно: а б с Фейгенбаум А., Мур Р., Кларк Дж., Хьюсон С., Читаят Д., Рэй П.Н., Стокли Т.Л. (январь 2004 г.). «Болезнь Канавана: определение частоты носительства у еврейского населения ашкенази и разработка нового молекулярного диагностического анализа». Американский журнал медицинской генетики. Часть А. 124А (2): 142–7. дои : 10.1002/ajmg.a.20334 . ПМИД 14699612 . S2CID 25981659 .
- ^ Перейти обратно: а б с д и Раб РМ, Михалс-Слейв К (март 2000 г.). «Губчатая дегенерация головного мозга, болезнь Канавана: биохимические и молекулярные данные» . Границы бионауки . 5 : Д307-11. дои : 10.2741/матлон . ПМИД 10704428 .
- ^ Перейти обратно: а б Мадхаварао К.Н., Арун П., Моффетт Дж.Р., Шукс С., Сурендран С., Маталон Р. и др. (апрель 2005 г.). «Нарушение катаболизма N-ацетиласпартата снижает уровень ацетата в мозге и синтез липидов миелина при болезни Канавана» . Труды Национальной академии наук Соединенных Штатов Америки . 102 (14): 5221–6. Бибкод : 2005PNAS..102.5221M . дои : 10.1073/pnas.0409184102 . ПМК 555036 . ПМИД 15784740 .
- ^ Сурендран С., Михалс-Маталон К., Кваст М.Дж., Тайринг С.К., Вэй Дж., Эзелль Э.Л., Маталон Р. (март 2006 г.). «Болезнь Канавана: моногенный признак со сложным геномным взаимодействием». Молекулярная генетика и обмен веществ . 80 (1–2): 74–80. дои : 10.1016/j.ymgme.2003.08.015 . ПМИД 14567959 .
- ^ Перейти обратно: а б с д и ж г час я дж к л м н тот Ахмед СС, Гао Г (2014). Зшоке Дж., Баумгартнер М., Морава Э., Паттерсон М. (ред.). «Придание белому веществу значения: прогресс в понимании болезни Канавана и терапевтических мер за восемь десятилетий» . Отчеты JIMD . 19 . Берлин, Гейдельберг: Springer Berlin Heidelberg: 11–22. дои : 10.1007/8904_2014_356 . ISBN 978-3-662-46189-1 . ПМК 4501231 . ПМИД 25604619 .
- ^ Маталон Р., Михалс К., Каул Р. (октябрь 1995 г.). «Болезнь Канавана: от губчатой дегенерации к молекулярному анализу». Журнал педиатрии . 127 (4): 511–7. дои : 10.1016/S0022-3476(95)70105-2 . ПМИД 7562269 .
- ^ Перейти обратно: а б с д Бухари М.Р., Саманта Д., Бухари С.Р. (2021). Болезнь Канавана . Остров сокровищ (Флорида): StatPearls Publishing. ПМИД 28613566 .
{{cite book}}:|work=игнорируется ( помогите ) - ^ Перейти обратно: а б Маталон Р., Дельгадо Л., Михалс-Маталон К. (январь 2020 г.). «Глава 66 - Болезнь Канавана». Розенберг Р.Н., Паскуаль Дж.М. (ред.). Молекулярные и генетические основы неврологических и психиатрических заболеваний Розенберга (шестое изд.). Академическая пресса. стр. 909–916. ISBN 978-0-12-813955-4 .
- ^ Перейти обратно: а б с д и ж Адачи М., Шнек Л., Кара Дж., Волк Б.В. (сентябрь 1973 г.). «Губчатая дегенерация центральной нервной системы (тип Ван Богерта и Бертрана; болезнь Канавана). Обзор». Патология человека . 4 (3): 331–47. дои : 10.1016/s0046-8177(73)80098-x . ПМИД 4593851 .
- ^ Перейти обратно: а б с Маталон Р., Тонкий Л., Михалс-Маталон К. (1993). Адам М.П., Ардингер Х.Х., Пагон Р.А., Уоллес С.Е., Бин Л.Дж., Мирза Г., Амемия А. (ред.). Болезнь Канавана . Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл. ПМИД 20301412 .
{{cite book}}:|work=игнорируется ( помогите ) - ^ Кантор Б., МакКаун Т., Леоне П., Грей С.Дж. (01 января 2014 г.). Клинические применения, связанные с переносом генов ЦНС . Достижения генетики. Том. 87. стр. 71–124. дои : 10.1016/B978-0-12-800149-3.00002-0 . ISBN 9780128001493 . ПМЦ 4518844 . ПМИД 25311921 .
- ^ Трегер ЕС, Рапин I (март 1998 г.). «Клиническое течение болезни Канавана». Детская неврология . 18 (3): 207–12. дои : 10.1016/s0887-8994(97)00185-9 . ПМИД 9568915 .
- ^ «Болезнь Канавана – MeSH – NCBI» . www.ncbi.nlm.nih.gov . Проверено 13 апреля 2021 г.
- ^ Цойтен Т (2000). «Молекулярные водяные насосы». Обзоры физиологии, биохимии и фармакологии . 141 . Берлин, Гейдельберг: Springer Berlin Heidelberg: 97–151. дои : 10.1007/bfb0119578 . ISBN 978-3-540-66627-1 . ПМИД 10916424 .
- ^ «Мозг - Внутримиелиновый отек - Атлас неопухолевых поражений» . ntp.niehs.nih.gov . Проверено 13 апреля 2021 г.
- ^ Перейти обратно: а б Ван Дж., Леоне П., Ву Дж., Фрэнсис Дж.С., Ли Х., Джайн М.Р. и др. (январь 2009 г.). «Нарушения миелиновых липидов у треморных крыс с дефицитом аспартоацилазы» . Нейрохимические исследования . 34 (1): 138–48. дои : 10.1007/s11064-008-9726-5 . ПМЦ 4640699 . ПМИД 18478328 .
- ^ Перейти обратно: а б Спандж С., Вагнер Т., Хайнцель Т., Кремер О.Г. (январь 2009 г.). «Ацетилирование негистоновых белков модулирует клеточную передачу сигналов на нескольких уровнях». Международный журнал биохимии и клеточной биологии . 41 (1): 185–98. doi : 10.1016/j.biocel.2008.08.027 . ПМИД 18804549 .
- ^ Кумар С., Бьянкотти Дж. К., Маталон Р., де Веллис Дж. (ноябрь 2009 г.). «Отсутствие активности аспартоацилазы нарушает выживаемость и дифференцировку нейрональных предшественников и олигодендроцитов в мышиной модели болезни Канавана». Журнал нейробиологических исследований . 87 (15): 3415–27. дои : 10.1002/jnr.22233 . ПМИД 19739253 . S2CID 44554774 .
- ^ Перейти обратно: а б «Дефицит аспартоацилазы (болезнь Канавана) | Метаболические и молекулярные основы наследственных заболеваний в Интернете | OMMBID | McGraw-Hill Medical» . ombbid.mhmedical.com . Проверено 15 апреля 2021 г.
- ^ Перейти обратно: а б Фрэнсис Дж.С., Странде Л., Марков В., Леоне П. (сентябрь 2012 г.). «Аспартоацилаза поддерживает окислительный энергетический обмен во время миелинизации» . Журнал церебрального кровотока и метаболизма . 32 (9): 1725–36. дои : 10.1038/jcbfm.2012.66 . ПМЦ 3434629 . ПМИД 22617649 .
- ^ Перейти обратно: а б Педерзолли С.Д., Меска С.П., Скапин Ф., Рокенбах Ф.Дж., Сгараватти А.М., Сгарби М.Б. и др. (август 2007 г.). «N-ацетиласпаргиновая кислота способствует окислительному стрессу в коре головного мозга крыс». Международный журнал нейробиологии развития . 25 (5): 317–24. doi : 10.1016/j.ijdevneu.2007.04.002 . ПМИД 17604935 . S2CID 41719397 .
- ^ Чуанг Ю.К. (март 2010 г.). «Митохондриальная дисфункция и окислительный стресс при гибели нейронов, вызванной судорогами» . Acta Neurologica Тайваньика . 19 (1): 3–15. ПМИД 20711885 .
- ^ Сринивасан П., Пурушотаман К.К. (январь 2013 г.). «Рентгенологический ключ к диагностике болезни Канавана». Индийский журнал педиатрии . 80 (1): 75–7. дои : 10.1007/ s12098-012-0794-9 ПМИД 22660905 . S2CID 35979708 .
- ^ «Болезнь Канавана, Анализ ДНК – Тесты – GTR – NCBI» . www.ncbi.nlm.nih.gov . Проверено 31 марта 2021 г.
- ^ Перейти обратно: а б «Информационная страница о болезни Канаван | Национальный институт неврологических расстройств и инсульта» . www.ninds.nih.gov . Проверено 31 марта 2021 г.
- ^ Перейти обратно: а б с Хосино Х., Кубота М. (август 2014 г.). «Болезнь Канавана: клинические особенности и последние достижения исследований» . Международная педиатрия . 56 (4): 477–83. дои : 10.1111/пед.12422 . ПМИД 24977939 . S2CID 206261733 .
- ^ Хант А., Бёрн Р. (январь 1995 г.). «Медико-сестринские проблемы детей с нейродегенеративными заболеваниями». Паллиативная медицина . 9 (1): 19–26. дои : 10.1177/026921639500900104 . ПМИД 7719515 . S2CID 20328927 .
- ^ Бэйкуэлл Дж. (август 2007 г.). «Выбор вспомогательного оборудования в детской терапии». Международный журнал терапии и реабилитации . 14 (8): 379–381. дои : 10.12968/ijtr.2007.14.8.24358 . ISSN 1741-1645 .
- ^ Перейти обратно: а б Роско Р.Б., Эллиотт С., Заррос А., Бэйли Г.С. (июль 2016 г.). «Негенетические терапевтические подходы к болезни Канавана» (PDF) . Журнал неврологических наук . 366 : 116–124. дои : 10.1016/j.jns.2016.05.012 . ПМИД 27288788 . S2CID 329155 .
- ^ Сурендран С., Бхатнагар М. (июнь 2011 г.). «Повышение уровня N-ацетиласпаргиновой кислоты вызывает окислительный стресс, внося свой вклад в патофизиологию заболевания». Международный журнал неврологии . 121 (6): 305–9. дои : 10.3109/00207454.2011.558225 . ПМИД 21348802 . S2CID 5406213 .
- ^ Мацунага С., Киши Т., Аннас П., Басун Х., Хампель Х., Ивата Н. (2015). «Литий как средство лечения болезни Альцгеймера: систематический обзор и метаанализ». Журнал болезни Альцгеймера . 48 (2): 403–10. дои : 10.3233/JAD-150437 . ПМИД 26402004 .
- ^ Линхард Ю, Сасс Дж (01 января 2011 г.). «Болезнь Канавана: нейрометаболическое заболевание, вызванное дефицитом аспартоацилазы» . Журнал педиатрических наук . 3 (1): 1–11. doi : 10.17334/jps.44686 (неактивен 31 января 2024 г.).
{{cite journal}}: CS1 maint: DOI неактивен по состоянию на январь 2024 г. ( ссылка ) - ^ Насо М.Ф., Томкович Б., Перри В.Л., Строл В.Р. (август 2017 г.). «Аденоассоциированный вирус (AAV) как вектор для генной терапии» . Биопрепараты . 31 (4): 317–334. дои : 10.1007/s40259-017-0234-5 . ПМЦ 5548848 . ПМИД 28669112 .
- ^ Ван Д., Тай П.В., Гао Г. (май 2019 г.). «Адено-ассоциированный вирусный вектор как платформа для доставки генной терапии» . Обзоры природы. Открытие наркотиков . 18 (5): 358–378. дои : 10.1038/s41573-019-0012-9 . ПМК 6927556 . ПМИД 30710128 .
- ^ Лю Д., Чжу М., Чжан Ю., Дяо Ю. (январь 2021 г.). «Пересечение гематоэнцефалического барьера векторами AAV». Метаболические заболевания головного мозга . 36 (1): 45–52. дои : 10.1007/s11011-020-00630-2 . ISSN 0885-7490 . ПМИД 33201426 . S2CID 226986888 .
- ^ Чен В., Ху Ю, Цзюй Д. (август 2020 г.). «Генная терапия нейродегенеративных заболеваний: достижения, идеи и перспективы» . Акта Фармасьютика Синика Б. 10 (8): 1347–1359. дои : 10.1016/j.apsb.2020.01.015 . ПМЦ 7488363 . ПМИД 32963936 .
- ^ Бас Н.Э., Дебросс С.Д. (1 января 2014 г.). «Болезнь Канавана» . Энциклопедия неврологических наук . Академическая пресса. стр. 577–579. дои : 10.1016/B978-0-12-385157-4.00088-9 . ISBN 9780123851581 .
- ^ Перейти обратно: а б Каул Р., Гао Г.П., Алоя М., Баламуруган К., Петроски А., Михалс К., Маталон Р. (июль 1994 г.). «Болезнь Канавана: мутации среди пациентов-евреев и неевреев» . Американский журнал генетики человека . 55 (1): 34–41. ПМК 1918221 . ПМИД 8023850 .
- ^ Глобус Дж. Х., Штраус I (1 декабря 1928 г.). «Прогрессирующая дегенеративная подкорковая энцефалопатия» . Архивы неврологии и психиатрии . 20 (6): 1190–1228. doi : 10.1001/archneurpsyc.1928.02210180041003 . ISSN 0096-6754 .
- ^ Клугманн, Матиас; Лейхтлейн, Клаудия Б. (2006). «Клинические испытания генной терапии болезни Канавана - I. Нозология болезни Канавана». В Каплитте, Майкл Г.; Во время, Мэтью Дж. (ред.). Генная терапия центральной нервной системы . стр. 303–316. дои : 10.1016/B978-012397632-1/50024-1 . ISBN 978-0-12-397632-1 .
- ^ Берторини, Тулио Э.; Перес, Ангел (2014). «Неврологические осложнения заболеваний надпочечников». В Биллере, Хосе; Ферро, Хосе М. (ред.). Справочник по клинической неврологии: неврологические аспекты системных заболеваний, часть II . Том. 120. стр. 749–771. дои : 10.1016/B978-0-7020-4087-0.00050-4 . ISBN 9780702040870 . ПМИД 24365350 .
- ^ Позер, СМ; Гутьер, Ф; Карпентье, Массачусетс; Айкарди, Дж. (январь 1986 г.). «Миелинокластический диффузный склероз Шильдера». Педиатрия . 77 (1): 107–112. ПМИД 3940347 . Опечатка в: Педиатрия . 78 (1): 138 (июль 1986 г.).
Термин «болезнь Шильдера» использовался для описания столь различных состояний, как адренолейкодистрофия, миелинокластический диффузный склероз... Эпонимическое обозначение следует зарезервировать для случаев миелинокластического диффузного склероза, соответствующих случаю, описанному Шильдером в 1912 году.
- ^ Канаван М.М. (февраль 1931 г.). «Диффуза периаксиального энцефалита Шильдера: сообщение о случае у ребенка в возрасте шестнадцати с половиной месяцев». Архивы неврологии и психиатрии . 25 (2): 299–308. doi : 10.1001/archneurpsyc.1931.02230020085005 .
- ^ Перейти обратно: а б с Адачи М (1967). «Исследования губчатой дегенерации центральной нервной системы (тип Ван Богерта – Бертрана)». Врожденные нарушения обмена сфинголипидов . Эльзевир. стр. 129–147. дои : 10.1016/b978-1-4831-9855-2.50013-7 . ISBN 978-1-4831-9855-2 .
- ^ Айсельсберг Ф (1 июня 1937 г.). «О семейном диффузном склерозе головного мозга раннего детства» . Журнал педиатрии (на немецком языке). 58 (6): 702–725. дои : 10.1007/BF02249721 . ISSN 1432-1076 . S2CID 31242183 .