Синдром Меккеля-Грубера
| синдром Меккеля | |
|---|---|
| Другие имена | Синдром Меккеля-Грубера , синдром Грубера , Dysencephalia splanchnocystica |
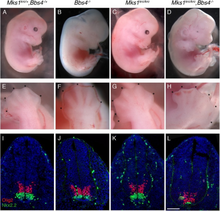 | |
| Эмбрионы с мутацией MKS1KRC, причиной синдрома Меккеля. | |
| Специальность | Медицинская генетика |
| Назван в честь |
|
Синдром Меккеля-Грубера — редкое летальное цилиопатическое генетическое заболевание , характеризующееся почек кистозной дисплазией , пороками развития центральной нервной системы (затылочное энцефалоцеле), полидактилией (постаксиальной), дефектами развития печени и гипоплазией легких вследствие маловодия . [ нужна ссылка ] Синдром Меккеля-Грубера назван в честь Иоганна Меккеля и Георга Грубера. [ 1 ] [ 2 ] [ 3 ]
Патофизиология
[ редактировать ]Синдром Меккеля-Грубера (МКС) — аутосомно-рецессивный летальный порок развития. Недавно два гена MKS : MKS1 и MKS3 были идентифицированы проведенное исследование описало клеточные , субклеточные и функциональные характеристики новых белков MKS1 . Недавно и меккелина , кодируемых этими генами. [ 4 ] Причиной этого смертельного заболевания является главным образом нарушение выработки этого белка. [ нужна ссылка ]
| Тип | МОЙ БОГ | Ген |
|---|---|---|
| МКС1 | 609883 | МКС1 |
| МКС2 | 603194 | ТМЕМ216 |
| МКС3 | 607361 | ТМЕМ67 |
| МКС4 | 611134 | CEP290 |
| МКС5 | 611561 | РПГРИП1Л |
| МКС6 | 612284 | CC2D2A |
| МКС7 | 608002 | НПХП3 |
| МКС8 | 613846 | ТКТН2 |
| МКС9 | 614144 | Б9Д1 |
| MKS10 | 611951 | Б9Д2 |
Связь с другими редкими генетическими заболеваниями
[ редактировать ]Недавние результаты генетических исследований показали, что большое количество генетических нарушений , как генетических синдромов , так и генетических заболеваний , которые ранее не были идентифицированы в медицинской литературе как родственные, на самом деле могут быть тесно связаны в генотипической первопричине широко распространенной патологии. различные фенотипически наблюдаемые расстройства . Таким образом, синдром Меккеля-Грубера представляет собой цилиопатию . Другие известные цилиопатии включают первичную цилиарную дискинезию , синдром Барде-Бидля , поликистоз почек и печени , нефронофтиз , синдром Альстрема и некоторые формы дегенерации сетчатки . [ 5 ] Ген MKS1 был идентифицирован как связанный с цилиопатией. [ 6 ]
Диагностика
[ редактировать ]Диспластические почки встречаются более чем в 95% всех выявленных случаев. Когда это происходит, внутри почки развиваются микроскопические кисты , которые медленно разрушают ее, в результате чего она увеличивается в 10–20 раз по сравнению с первоначальным размером. Уровень околоплодных вод в матке может значительно изменяться или оставаться нормальным, и нормальный уровень жидкости не должен быть критерием исключения диагноза. [ нужна ссылка ]
Затылочное энцефалоцеле присутствует в 60–80% всех случаев, а постаксиальная полидактилия – в 55–75% от общего числа выявленных случаев. Также часто наблюдаются искривление или укорочение конечностей. [ нужна ссылка ]
Обнаружение хотя бы двух из трех фенотипических особенностей классической триады при наличии нормального кариотипа делает диагноз достоверным. Регулярное УЗИ и превентивный дородовой уход обычно позволяют обнаружить симптомы на ранних стадиях беременности . [ нужна ссылка ]
Управление
[ редактировать ]Лекарства от этой болезни не существует. Лечение симптоматическое и направлено на то, чтобы малышу было максимально комфортно. [ 7 ]
Прогноз
[ редактировать ]Болезнь смертельна. Большинство младенцев, которые не рождаются мертворожденными с синдромом Меккеля, умирают в течение нескольких часов или дней после рождения. [ 8 ] Самый длительный период выживания, о котором сообщается в медицинской литературе, составляет 28 месяцев. [ 9 ]
Заболеваемость
[ редактировать ]Хотя это точно не известно, предполагается, что общий уровень заболеваемости , по данным Бергсмы, [ 10 ] для синдрома Меккеля составляет 0,02 на 10 000 рождений. По данным другого исследования, проведенного шесть лет спустя, уровень заболеваемости мог варьироваться от 0,07 до 0,7 на 10 000 рождений. [ 11 ]
Этот синдром является болезнью финского происхождения . Его частота намного выше в Финляндии , где заболеваемость достигает 1,1 на 10 000 рождений. Подсчитано, что синдром Меккеля составляет 5% всех дефектов нервной трубки . [ 12 ] Исследование перинатальной смертности в Лестершире за период с 1976 по 1982 год выявило высокий уровень заболеваемости. [ написание? ] синдрома Меккеля у иммигрантов из Индии из Гуджарата . [ 13 ]
Ссылки
[ редактировать ]- ^ Synd/2055 в Who Named It?
- ^ Дж. Ф. Меккель. Описание двух братьев и сестер, изуродованных очень похожими отклонениями в образовании. Немецкие архивы физиологии, 1822, 7 : 99–172.
- ^ ГБ Грубер. Вклад в вопрос о «связанных» пороках развития (акроцефалосиндактилия и дизенцефалия спланкноцистика. Бейтр путь Анат, 1934, 93 : 459–476.
- ^ Доу Х.Р., Смит У.М., Каллинейн А.Р., Джеррелли Д., Кокс П., Бадано Дж.Л., Блэр-Рид С., Шрирам Н., Катсанис Н., Атти-Битах Т., Аффорд СК, Копп А.Дж., Келли Д.А., Галл К., Джонсон Калифорния (2007 г.) ). «Белки синдрома Меккеля-Грубера MKS1 и меккелин взаимодействуют и необходимы для формирования первичных ресничек» . Молекулярная генетика человека . 16 (2): 173–186. дои : 10.1093/hmg/ddl459 . ПМИД 17185389 .
- ^ Бадано, Хосе Л.; Норимаса Мицума; Фил Л. Билз; Николас Кацанис (сентябрь 2006 г.). «Цилиопатии: новый класс генетических заболеваний человека». Ежегодный обзор геномики и генетики человека . 7 : 125–148. дои : 10.1146/annurev.genom.7.080505.115610 . ПМИД 16722803 .
- ^ Киттяля, Мира (май 2006 г.). «Идентификация гена синдрома Меккеля (MKS1) раскрывает новую цилиопатию» (PDF) . Национальный институт общественного здравоохранения, Хельсинки. Архивировано из оригинала (PDF) 21 июля 2006 г. Проверено 6 июля 2008 г.
{{cite journal}}: Для цитирования журнала требуется|journal=( помощь ) - ^ «Синдром Меккеля» . НОРД (Национальная организация по редким заболеваниям) . Проверено 2 декабря 2019 г.
- ^ Хейр, Абдель-Монейм ЭМ; Имам Абдель Муталаб; Омер, Ильхам М.; Хасан, Ибцама М.А.; Эламин, Сара А.; Авадалла, Эсра А.; Гадалла, Мохаммед Х.; Хамдун, Тагва А. (2012). «Синдром Меккеля-Грубера: редкая и смертельная аномалия» . Суданский педиатрический журнал . 12 (1): 93–96. ISSN 0256-4408 . ПМЦ 4949827 . ПМИД 27493335 .
- ^ Баришич, Ингеборг; Бобан, Любица; Лоан, Мария; Гарн, Эстер; Уэлсли, Диана; Кальцолари, Элиза; Долк, Хелен; Аддор, Мари-Клод; Бергман, Йорике Э.Х.; Браз, Паула; Дрейпер, Элизабет С. (июнь 2015 г.). «Синдром Меккеля-Грубера: популяционное исследование распространенности, пренатальной диагностики, клинических особенностей и выживаемости в Европе» . Европейский журнал генетики человека . 23 (6): 746–752. дои : 10.1038/ejhg.2014.174 . ISSN 1018-4813 . ПМК 4795048 . ПМИД 25182137 .
- ^ Бергсма, Д. (1979). «Врожденные дефекты». Атлас и сборник . Лондон: Макмиллан Пресс.
- ^ Салонен, Р.; Норио, Р.; Рейнольдс, Джеймс Ф. (1984). «Синдром Меккеля: клинико-патологические данные у 67 пациентов». Американский журнал медицинской генетики . 18 (4): 671–689. дои : 10.1002/ajmg.1320180414 . ПМИД 6486167 .
- ^ Нюберг, Д.А.; и др. (1990). «Синдром Меккеля-Грубера; важность пренатальной диагностики». Журнал ультразвука в медицине . 9 (12): 691–696. дои : 10.7863/июм.1990.9.12.691 . ПМИД 2277397 . S2CID 25658017 .
- ^ Янг, ID; Рикетт, AB; Кларк, М. (1 августа 1985 г.). «Высокая заболеваемость синдромом Меккеля у индейцев Гуджарати» . Журнал медицинской генетики . 22 (4): 301–304. дои : 10.1136/jmg.22.4.301 . ISSN 0022-2593 . ПМЦ 1049454 . ПМИД 4045959 .