РНК-Seq

RNA-Seq (названный как сокращение от секвенирования РНК ) — это метод, который использует секвенирование следующего поколения для выявления присутствия и количества молекул РНК в биологическом образце, обеспечивая моментальный снимок экспрессии генов в образце, также известный как транскриптом . [2] [3]
В частности, RNA-Seq облегчает возможность изучения альтернативных транскриптов сплайсинга генов , посттранскрипционных модификаций , слияния генов , мутаций/ SNP и изменений в экспрессии генов с течением времени или различий в экспрессии генов в разных группах или методах лечения. [4] Помимо транскриптов мРНК, RNA-Seq может исследовать различные популяции РНК, включая общую РНК, малые РНК, такие как микроРНК , тРНК и профилирование рибосом . [5] RNA-Seq также можно использовать для определения границ экзонов и интронов , а также для проверки или изменения ранее аннотированных 5'- и 3'- границ генов. Последние достижения в области секвенирования РНК включают секвенирование отдельных клеток , массовое секвенирование РНК , [6] Секвенирование 3'-мРНК , секвенирование фиксированной ткани in situ и секвенирование нативных молекул РНК с секвенированием одиночных молекул в реальном времени. [7] Другими примерами новых приложений RNA-Seq благодаря развитию алгоритмов биоинформатики являются изменение числа копий, микробное загрязнение, мобильные элементы, тип клеток (деконволюция) и присутствие неоантигенов. [8]
До RNA-Seq исследования экспрессии генов проводились с помощью микрочипов на основе гибридизации . знания последовательности Проблемы с микрочипами включают артефакты перекрестной гибридизации, плохую количественную оценку генов с низкой и высокой экспрессией, а также необходимость априорного . [9] Из-за этих технических проблем транскриптомика перешла на методы, основанные на секвенировании. Они перешли от секвенирования по Сэнгеру библиотек меток экспрессируемых последовательностей к методам на основе химических меток (например, серийный анализ экспрессии генов ) и, наконец, к современной технологии — (кДНК) следующего поколения секвенированию комплементарной ДНК , особенно RNA-Seq.

Методы
[ редактировать ]Подготовка библиотеки
[ редактировать ]
Общие шаги по подготовке библиотеки комплементарной ДНК (кДНК) для секвенирования описаны ниже, но часто различаются в зависимости от платформы. [10] [3] [11]
- Выделение РНК: РНК выделяют из ткани и смешивают с дезоксирибонуклеазой (ДНКазой). ДНКаза уменьшает количество геномной ДНК. Степень деградации РНК проверяют с помощью гель- и капиллярного электрофореза и используют для присвоения показателя целостности РНК образцу . Качество РНК и общее количество исходной РНК учитываются на последующих этапах подготовки библиотеки, секвенирования и анализа.
- Селекция/истощение РНК: для анализа представляющих интерес сигналов выделенную РНК можно либо сохранить как есть, обогатив РНК с 3'-полиаденилированными (поли(А)) хвостами, чтобы включать только эукариотическую мРНК , обедненную рибосомальной РНК (рРНК) , и /или отфильтрован на предмет РНК, которая связывает определенные последовательности ( таблица методов отбора и истощения РНК ниже). Молекулы РНК, имеющие 3'-поли(А)-хвосты у эукариот, в основном состоят из зрелых, процессированных, кодирующих последовательностей. Отбор поли(А) осуществляется путем смешивания РНК с олигомерами поли(Т), ковалентно связанными с подложкой, обычно магнитными шариками. [12] [13] Селекция поли(А) имеет важные ограничения при определении биотипа РНК. Многие биотипы РНК не являются полиаденилированными, включая многие некодирующие РНК и транскрипты белков ядра гистонов, или регулируются посредством длины их поли(А)-хвоста (например, цитокины) и, таким образом, не могут быть обнаружены после селекции поли(А). [14] Более того, селекция поли(А) может демонстрировать повышенную 3'-смещенность, особенно с РНК более низкого качества. [15] [16] Этих ограничений можно избежать с помощью истощения рибосом, удаляя рРНК, которая обычно составляет более 90% РНК в клетке. Как обогащение поли(А), так и этапы истощения рибосом являются трудоемкими и могут привести к систематическим ошибкам, поэтому были разработаны более простые подходы для пропуска этих этапов. [17] Малые РНК-мишени, такие как микроРНК , могут быть дополнительно выделены путем выбора размера с помощью эксклюзивных гелей, магнитных шариков или коммерческих наборов.
- Синтез кДНК: РНК обратно транскрибируется в кДНК, поскольку ДНК более стабильна и позволяет проводить амплификацию (в которой используются ДНК-полимеразы ) и использовать более зрелую технологию секвенирования ДНК. Амплификация после обратной транскрипции приводит к потере многоцепочности , чего можно избежать с помощью химического мечения или секвенирования одной молекулы. Фрагментация и выбор размера выполняются для очистки последовательностей, длина которых соответствует длине секвенатора. РНК, кДНК или обе фрагментируются с помощью ферментов, обработки ультразвуком , двухвалентными ионами или распылителями. Фрагментация РНК уменьшает 5'-смещение случайной обратной транскрипции и влияние сайтов связывания праймеров . [13] с тем недостатком, что 5'- и 3'-концы менее эффективно преобразуются в ДНК. За фрагментацией следует выбор размера, при котором либо удаляются небольшие последовательности, либо выбирается ограниченный диапазон длин последовательностей. Поскольку малые РНК, такие как микроРНК, теряются, их анализируют независимо. кДНК для каждого эксперимента может быть проиндексирована гексамерным или октамерным штрих-кодом, так что эти эксперименты можно объединить в одну дорожку для мультиплексного секвенирования.
| Стратегия | Преобладающий тип РНК | Содержание рибосомальной РНК | Необработанное содержимое РНК | Метод изоляции |
|---|---|---|---|---|
| Общая РНК | Все | Высокий | Высокий | Никто |
| PolyA selection | Кодирование | Низкий | Низкий | Гибридизация поли(дТ) с олигомерами |
| истощение рРНК | Кодирование, некодирование | Низкий | Высокий | Удаление олигомеров, комплементарных рРНК |
| захват РНК | Целевой | Низкий | Умеренный | Гибридизация с зондами, комплементарными желаемым транскриптам |
Комплементарное секвенирование ДНК (cDNA-Seq)
[ редактировать ]Библиотека кДНК, полученная из биотипов РНК, затем секвенируется в формат, читаемый компьютером. Существует множество высокопроизводительных технологий секвенирования кДНК, включая платформы, разработанные Illumina , Thermo Fisher , BGI/MGI , PacBio и Oxford Nanopore Technologies . [18] При секвенировании короткого считывания Illumina, распространенной технологии секвенирования кДНК, адаптеры лигируются с кДНК, ДНК прикрепляется к проточной ячейке, кластеры генерируются посредством циклов мостиковой амплификации и денатурации, а синтез последовательности выполняется циклами. синтеза комплементарных цепей и лазерного возбуждения оснований с обратимыми терминаторами. Выбор и параметры платформы секвенирования определяются дизайном и стоимостью эксперимента. Общие соображения по планированию эксперимента включают принятие решения о длине секвенирования, глубине секвенирования, использовании одноконцевого или парного секвенирования, количестве повторов, мультиплексировании, рандомизации и всплесках. [19]
Секвенирование малых РНК/некодирующих РНК
[ редактировать ]При секвенировании РНК, отличной от мРНК, препарат библиотеки модифицируют. Клеточная РНК выбирается на основе желаемого диапазона размеров. Для небольших мишеней РНК, таких как микроРНК , РНК выделяют путем выбора размера. Это можно выполнить с помощью геля для исключения размера, с помощью магнитных шариков для выбора размера или с помощью коммерчески разработанного набора. После выделения линкеры добавляют к 3'- и 5'-концам, а затем очищают. Последним шагом является генерация кДНК посредством обратной транскрипции.
Прямое секвенирование РНК
[ редактировать ]
Поскольку было показано, что преобразование РНК в кДНК , лигирование, амплификация и другие манипуляции с образцами приводят к искажениям и артефактам, которые могут мешать как правильной характеристике, так и количественной оценке транскриптов, [20] Прямое секвенирование одной молекулы РНК исследовалось такими компаниями, как Helicos (банкрот), Oxford Nanopore Technologies , [21] и другие. Эта технология секвенирует молекулы РНК напрямую и массово-параллельно.
Секвенирование одной молекулы РНК в реальном времени
[ редактировать ]Массивно-параллельное прямое секвенирование РНК с одной молекулой было исследовано как альтернатива традиционному секвенированию РНК, при котором преобразование РНК в кДНК , лигирование, амплификация и другие этапы манипуляций с образцами могут приводить к систематическим ошибкам и артефактам. [22] Технологические платформы, которые выполняют секвенирование РНК одной молекулы в реальном времени, включают Oxford Nanopore Technologies (ONT) секвенирование нанопор , [21] PacBio IsoSeq и Helicos (банкрот). Секвенирование РНК в ее нативной форме сохраняет такие модификации, как метилирование, что позволяет исследовать их напрямую и одновременно. [21] Еще одним преимуществом одномолекулярного RNA-Seq является то, что транскрипты могут быть охвачены по всей длине, что позволяет более уверенно обнаруживать и количественно определять изоформы по сравнению с секвенированием с коротким считыванием. Традиционно методы одномолекулярного RNA-Seq имеют более высокий уровень ошибок по сравнению с секвенированием с коротким считыванием, но новые методы, такие как ONT, позволяют напрямую RNA-Seq ограничивать ошибки, избегая фрагментации и конверсии кДНК. Недавнее использование прямого секвенирования РНК ONT для дифференциальной экспрессии в популяциях клеток человека продемонстрировало, что эта технология может преодолеть многие ограничения секвенирования коротких и длинных кДНК. [23]
Секвенирование одноклеточной РНК (scRNA-Seq)
[ редактировать ]Стандартные методы, такие как микрочипы и стандартный анализ объемной РНК-Seq, анализируют экспрессию РНК из больших популяций клеток. В смешанных популяциях клеток эти измерения могут скрывать критические различия между отдельными клетками внутри этих популяций. [24] [25]
Секвенирование одноклеточной РНК (scRNA-Seq) позволяет получить профили экспрессии отдельных клеток. Хотя невозможно получить полную информацию о каждой РНК, экспрессируемой каждой клеткой, из-за небольшого количества доступного материала, закономерности экспрессии генов можно определить с помощью анализа кластеризации генов . Это может раскрыть существование редких типов клеток в клеточной популяции, которые, возможно, никогда раньше не наблюдались. Например, редкие специализированные клетки в легких, называемые легочными ионоцитами , которые экспрессируют регулятор трансмембранной проводимости при муковисцидозе, были идентифицированы в 2018 году двумя группами, выполнявшими scRNA-Seq на эпителии дыхательных путей легких. [26] [27]
Экспериментальные процедуры
[ редактировать ]
Текущие протоколы scRNA-Seq включают следующие этапы: выделение отдельной клетки и РНК, обратную транскрипцию (RT), амплификацию, создание библиотеки и секвенирование. Отдельные клетки либо механически разделяют в микролунки (например, BD Rhapsody, Takara ICELL8, Vycap Puncher Platform или CellMicrosystems CellRaft), либо инкапсулируют в капли (например, 10x Genomics Chromium, Illumina Bio-Rad ddSEQ, 1CellBio InDrop, Dolomite Bio Nadia). [28] Отдельные клетки метят путем добавления шариков со штрих-кодированными олигонуклеотидами; как клетки, так и шарики поставляются в ограниченных количествах, так что совместное заселение несколькими клетками и шариками является очень редким явлением. После завершения обратной транскрипции кДНК из многих клеток можно смешать для секвенирования; транскрипты из конкретной клетки идентифицируются уникальным штрих-кодом каждой клетки. [29] [30] Уникальный молекулярный идентификатор (UMI) может быть прикреплен к целевым последовательностям мРНК/кДНК, чтобы помочь идентифицировать артефакты во время подготовки библиотеки. [31]
Задачи scRNA-Seq включают сохранение первоначального относительного содержания мРНК в клетке и выявление редких транскриптов. [32] Этап обратной транскрипции имеет решающее значение, поскольку эффективность реакции RT определяет, какая часть популяции РНК клетки в конечном итоге будет проанализирована секвенатором. Процессивность обратных транскриптаз и используемые стратегии прайминга могут влиять на производство полноразмерной кДНК и создание библиотек, смещенных к 3'- или 5'-концу генов.
На этапе амплификации in vitro в настоящее время для амплификации кДНК используется либо ПЦР, либо транскрипция (IVT). Одним из преимуществ методов на основе ПЦР является возможность генерировать полноразмерную кДНК. Однако различная эффективность ПЦР для определенных последовательностей (например, содержания GC и структуры snapback) также может экспоненциально усиливаться, создавая библиотеки с неравномерным покрытием. С другой стороны, хотя библиотеки, созданные с помощью IVT, могут избежать смещения последовательностей, вызванного ПЦР, определенные последовательности могут транскрибироваться неэффективно, что приводит к выпадению последовательностей или образованию неполных последовательностей. [33] [24] Было опубликовано несколько протоколов scRNA-Seq:Тан и др., [34] СТРТ, [35] SMART-последовательность, [36] CEL-сек, [37] RAGE-сиквел, [38] Кварц-сек [39] и C1-КЛЕТКА. [40] Эти протоколы различаются стратегиями обратной транскрипции, синтеза и амплификации кДНК, а также возможностью использования штрих-кодов, специфичных для последовательности (т. е. UMI ), или способностью обрабатывать объединенные образцы. [41]
В 2017 году были представлены два подхода для одновременного измерения одноклеточной экспрессии мРНК и белка с помощью меченных олигонуклеотидами антител, известных как REAP-seq: [42] и CITE-сек. [43]
Приложения
[ редактировать ]scRNA-Seq становится широко используемым в биологических дисциплинах, включая развитие, неврологию , [44] онкология , [45] [46] [47] Аутоиммунное заболевание , [48] и Инфекционные заболевания . [49]
scRNA-Seq предоставил значительную информацию о развитии эмбрионов и организмов, включая червя Caenorhabditis elegans , [50] и регенеративная планария Schmidtea mediterranea . [51] [52] Первыми позвоночными животными, картографированными таким образом, были рыбки данио. [53] [54] и Xenopus laevis . [55] В каждом случае изучались несколько стадий эмбриона, что позволяло картировать весь процесс развития на клеточной основе. [10] Наука признала эти достижения « Прорывом года 2018» . [56]
Экспериментальные соображения
[ редактировать ]различные параметры При планировании и проведении экспериментов по секвенированию РНК учитываются :
- Специфичность ткани: экспрессия генов варьируется внутри и между тканями, и RNA-Seq измеряет это сочетание типов клеток. Это может затруднить выделение интересующего биологического механизма. Секвенирование отдельных клеток можно использовать для изучения каждой клетки по отдельности, что позволяет решить эту проблему.
- Зависимость от времени: экспрессия генов меняется со временем, и RNA-Seq делает только моментальный снимок. Можно провести эксперименты с временной динамикой, чтобы наблюдать изменения в транскриптоме.
- Охват (также известный как глубина): РНК содержит те же мутации, что и ДНК, и их обнаружение требует более глубокого охвата. При достаточно высоком охвате RNA-Seq можно использовать для оценки экспрессии каждой аллели. Это может дать представление о таких явлениях, как импринтинг или цис-регуляторные эффекты . Глубину секвенирования, необходимую для конкретных приложений, можно экстраполировать на основе пилотного эксперимента. [57]
- Артефакты генерации данных (также известные как технические отклонения): реагенты (например, набор для подготовки библиотеки), задействованный персонал и тип секвенатора (например, Illumina , Pacific Biosciences ) могут привести к техническим артефактам, которые могут быть ошибочно интерпретированы как значимые результаты. . Как и в случае любого научного эксперимента, разумно проводить секвенирование РНК в хорошо контролируемых условиях. Если это невозможно или исследование представляет собой метаанализ , другим решением является обнаружение технических артефактов путем определения скрытых переменных (обычно анализ главных компонентов или факторный анализ ) и последующей коррекции этих переменных. [58]
- Управление данными. Один эксперимент RNA-Seq на людях обычно занимает 1–5 ГБ (в сжатом виде) или более, если включать промежуточные файлы. [59] Такой большой объем данных может вызвать проблемы с хранением. Одним из решений является сжатие данных с использованием многоцелевых вычислительных схем (например, gzip ) или схем, специфичных для геномики. Последние могут быть основаны на эталонных последовательностях или de novo. Другим решением является проведение экспериментов на микрочипах, которых может быть достаточно для работы, основанной на гипотезах, или повторных исследований (в отличие от поисковых исследований).
Анализ
[ редактировать ]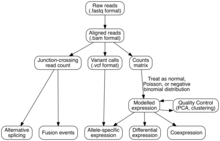
Сборка транскриптома
[ редактировать ]Для сопоставления считываний необработанной последовательности с геномными особенностями (т. е. сборки транскриптома) используются два метода:
- De novo: этот подход не требует эталонного генома для реконструкции транскриптома и обычно используется, если геном неизвестен, неполный или существенно изменен по сравнению с эталонным. [60] Проблемы при использовании коротких чтений для сборки de novo включают 1) определение того, какие чтения должны быть объединены в непрерывные последовательности ( контиги ), 2) устойчивость к ошибкам секвенирования и другим артефактам и 3) вычислительную эффективность. Основной алгоритм, используемый для сборки de novo, перешел от графов перекрытия, которые идентифицируют все парные перекрытия между чтениями, к графам де Брёйна , которые разбивают чтения на последовательности длины k и сжимают все k-меры в хеш-таблицу. [61] Графики перекрытия использовались при секвенировании по Сэнгеру, но они плохо масштабируются для миллионов прочтений, полученных с помощью RNA-Seq. Примерами ассемблеров, использующих графы де Брёйна, являются Trinity, [60] Оазисы [62] (получено из ассемблера генома Velvet [63] ), Бриджер, [64] и рнаСПАдес. [65] Секвенирование парного конца и длинного считывания одного и того же образца может устранить недостатки секвенирования короткого считывания, выступая в качестве шаблона или скелета. Метрики для оценки качества сборки de novo включают медианную длину контигов, количество контигов и N50 . [66]
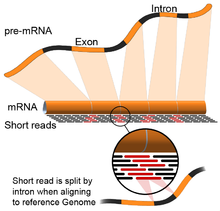
- Управление геномом: этот подход основан на тех же методах, которые используются для выравнивания ДНК, но с дополнительной сложностью выравнивания считываний, которые охватывают прерывистые части эталонного генома. [67] Эти прерывистые чтения являются результатом секвенирования сплайсированных транскриптов (см. рисунок). Обычно алгоритмы выравнивания состоят из двух этапов: 1) выравнивание коротких частей считывания (т. е. заполнение генома) и 2) использование динамического программирования для поиска оптимального выравнивания, иногда в сочетании с известными аннотациями. Программные инструменты, использующие выравнивание на основе генома, включают Bowtie , [68] TopHat (который использует результаты BowTie для выравнивания мест сращивания), [69] [70] Подчтение, [71] ЗВЕЗДА, [67] ХИСАТ2, [72] и ГМАП. [73] Результаты инструментов выравнивания (картирования) генома могут в дальнейшем использоваться такими инструментами, как Cufflinks. [70] или StringTie [74] для реконструкции смежных последовательностей транскриптов ( т.е. файла FASTA). Качество сборки, управляемой геномом, можно измерить как с помощью 1) показателей сборки de novo (например, N50), так и 2) сравнения с известными последовательностями транскрипта, сплайсингового соединения, генома и белка с использованием точности, отзыва или их комбинации (например, Оценка F1). [66] Кроме того, оценка in silico может быть выполнена с использованием моделирования чтения. [75] [76]
Примечание о качестве сборки: в настоящее время существует общее мнение, что 1) качество сборки может варьироваться в зависимости от используемого показателя, 2) инструменты сборки, которые получили хорошие оценки у одного вида, не обязательно хорошо работают у другого вида, и 3) сочетание разных подходов. может быть самым надежным. [77] [78] [79]
Количественная оценка экспрессии генов
[ редактировать ]Экспрессия количественно оценивается для изучения клеточных изменений в ответ на внешние раздражители, различий между здоровыми и больными состояниями, а также других исследовательских вопросов. Уровни транскриптов часто используются в качестве показателя количества белков, но они часто не эквивалентны из-за посттранскрипционных событий, таких как интерференция РНК и нонсенс-опосредованный распад . [80]
Экспрессию определяют количественно путем подсчета количества прочтений, сопоставленных с каждым локусом на этапе сборки транскриптома . Экспрессию экзонов или генов можно оценить количественно с использованием контигов или аннотаций эталонных транскриптов. [10] Эти наблюдаемые количества считываний RNA-Seq были надежно подтверждены более старыми технологиями, включая экспрессионные микрочипы и qPCR . [57] [81] Инструменты, определяющие количество: HTSeq, [82] Количество функций, [83] Раунт, [84] максимальное количество, [85] ФИКСЕК, [86] и Каффвант. Эти инструменты определяют количество чтений на основе выровненных данных RNA-Seq, но подсчеты без выравнивания также можно получить с помощью Sailfish. [87] and Kallisto. [88] Затем количество прочтений преобразуется в соответствующие показатели для проверки гипотез, регрессии и другого анализа. Параметры для этого преобразования:
- Глубина/охват секвенирования . Хотя глубина секвенирования заранее указывается при проведении нескольких экспериментов по секвенированию РНК, она все равно будет сильно различаться между экспериментами. [89] Таким образом, общее количество чтений, сгенерированных в одном эксперименте, обычно нормализуется путем преобразования количества во фрагменты, чтения или количества на миллион сопоставленных чтений (FPM, RPM или CPM). Разница между RPM и FPM исторически возникла в ходе эволюции от одноконцевого секвенирования фрагментов к парному секвенированию. При одноконцевом секвенировании на каждый фрагмент выполняется только одно чтение ( т. е . RPM = FPM). При секвенировании парных концов на каждый фрагмент приходится два чтения ( т. е . RPM = 2 x FPM). Глубину секвенирования иногда называют размером библиотеки — количеством промежуточных молекул кДНК в эксперименте.
- Длина гена: более длинные гены будут иметь больше фрагментов/чтений/счетов, чем более короткие гены, если экспрессия транскриптов одинакова. Это корректируется путем деления FPM на длину признака (который может быть геном, транскриптом или экзоном), в результате чего получается количество метрических фрагментов на тысячу оснований признака на миллион картированных чтений (FPKM). [90] При просмотре групп функций в выборках FPKM преобразуется в количество транскриптов на миллион (TPM) путем деления каждой FPKM на сумму FPKM в выборке. [91] [92] [93]
- Общий выход РНК образца: поскольку из каждого образца экстрагируется одинаковое количество РНК, образцы с большим количеством общей РНК будут содержать меньше РНК на ген. Эти гены, по-видимому, имеют пониженную экспрессию, что приводит к ложноположительным результатам последующих анализов. [89] Стратегии нормализации, включая квантиль, DESeq2, TMM и медианное соотношение, пытаются учесть эту разницу путем сравнения набора недифференцированно экспрессируемых генов между образцами и соответствующего масштабирования. [94]
- Дисперсия экспрессии каждого гена: моделируется для учета ошибки выборки (важно для генов с низким количеством считываний), увеличения мощности и уменьшения ложноположительных результатов. Дисперсию можно оценить как нормальное , Пуассоновое или отрицательное биномиальное распределение. [95] [96] [97] и часто разлагается на технические и биологические вариации.
Всплески для абсолютного количественного определения и обнаружения общегеномных эффектов
[ редактировать ]Вставки РНК представляют собой образцы РНК в известных концентрациях, которые можно использовать в качестве золотых стандартов при разработке экспериментов и во время последующих анализов для абсолютного количественного определения и обнаружения общегеномных эффектов.
- Абсолютная количественная оценка: Абсолютная количественная оценка экспрессии генов невозможна в большинстве экспериментов RNA-Seq, которые количественно определяют экспрессию относительно всех транскриптов. Это возможно путем выполнения RNA-Seq с добавлением образцов РНК в известных концентрациях. После секвенирования количество считываний вставных последовательностей используется для определения взаимосвязи между количеством считываний каждого гена и абсолютным количеством биологических фрагментов. [13] [98] В одном примере этот метод использовался на эмбрионах Xenopus тропического для определения кинетики транскрипции. [99]
- Обнаружение общегеномных эффектов: изменения в глобальных регуляторах, включая ремоделеры хроматина , факторы транскрипции (например, MYC ), ацетилтрансферазные комплексы и расположение нуклеосом, не соответствуют предположениям о нормализации, и контроль всплеска может предложить точную интерпретацию. [100] [101]
Дифференциальное выражение
[ редактировать ]Самое простое, но зачастую наиболее эффективное применение RNA-Seq — это обнаружение различий в экспрессии генов между двумя или более состояниями ( например , леченными и нелеченными); этот процесс называется дифференциальным выражением. Выходные данные часто называют дифференциально экспрессируемыми генами (DEG), и эти гены могут иметь повышенную или пониженную регуляцию ( т.е. повышенную или пониженную регуляцию в интересующем состоянии). Существует множество инструментов, выполняющих дифференциальное выражение . Большинство из них запускаются в R , Python или командной строке Unix . Обычно используемые инструменты включают DESeq, [96] крайР, [97] и воом+лимма, [95] [102] все это доступно через R/ Bioconductor . [103] [104] Вот общие соображения при выполнении дифференциального выражения:
- Входные данные: входные данные для дифференциальной экспрессии включают (1) матрицу экспрессии RNA-Seq (M генов x N образцов) и (2) матрицу дизайна, содержащую экспериментальные условия для N образцов. Простейшая матрица проектирования содержит один столбец, соответствующий меткам проверяемого условия. Другие ковариаты (также называемые факторами, признаками, метками или параметрами) могут включать пакетные эффекты , известные артефакты и любые метаданные, которые могут искажать или опосредовать экспрессию генов. Помимо известных ковариат, неизвестные ковариаты также могут быть оценены с помощью подходов неконтролируемого машинного обучения , включая главный компонент , суррогатную переменную, [105] и ПИР [58] анализы. Анализ скрытых переменных часто используется для данных секвенирования РНК тканей человека, которые обычно содержат дополнительные артефакты, не отраженные в метаданных ( например , время ишемии, источники из нескольких учреждений, основные клинические характеристики, сбор данных за многие годы с участием большого количества сотрудников).
- Методы: Большинство инструментов используют регрессию или непараметрическую статистику для идентификации дифференциально экспрессируемых генов и основаны либо на количестве чтений, сопоставленных с эталонным геномом (DESeq2, limma, EdgeR), либо на основе количества чтений, полученных в результате количественного анализа без выравнивания (sleuth, [106] Манжета, [107] бальное платье [108] ). [109] После регрессии большинство инструментов используют корректировки p-значения либо коэффициента семейных ошибок (FWER) , либо коэффициента ложных открытий (FDR) для учета нескольких гипотез (в исследованиях на людях ~ 20 000 генов, кодирующих белок, или ~ 50 000 биотипов).
- Выходные данные: Типичные выходные данные состоят из строк, соответствующих количеству генов, и как минимум трех столбцов, логарифмического изменения каждого гена ( логарифмическое преобразование соотношения экспрессии между условиями, меры размера эффекта ), p-значения и p. -значение скорректировано для множественных сравнений . Гены считаются биологически значимыми, если они проходят пороговые значения по величине эффекта (логарифмическое изменение) и статистической значимости . В идеале эти пороговые значения должны быть определены заранее , но характер экспериментов по секвенированию РНК часто носит исследовательский характер, поэтому трудно заранее предсказать величину эффекта и соответствующие пороговые значения.
- Подводные камни: Смысл существования этих сложных методов состоит в том, чтобы избежать множества ловушек, которые могут привести к статистическим ошибкам и вводящим в заблуждение интерпретациям. Ловушки включают повышенный уровень ложноположительных результатов (из-за множественных сравнений), артефакты при подготовке образцов, гетерогенность образцов (например, смешанный генетический фон), сильно коррелированные образцы, неучтенные многоуровневые экспериментальные планы и плохой экспериментальный дизайн . Одной из примечательных ошибок является просмотр результатов в Microsoft Excel без использования функции импорта, чтобы гарантировать, что имена генов останутся текстовыми. [110] Несмотря на удобство, Excel автоматически преобразует некоторые имена генов ( SEPT1 , DEC1 , MARCH2 ) в даты или числа с плавающей запятой.
- Выбор инструментов и сравнительное тестирование: существует множество попыток сравнить результаты этих инструментов, при этом DESeq2 имеет тенденцию умеренно превосходить другие методы. [111] [112] [113] [114] [19] [109] [115] [116] Как и в случае с другими методами, бенчмаркинг состоит из сравнения результатов инструмента друг с другом и с известными золотыми стандартами .
Последующий анализ списка дифференциально экспрессируемых генов бывает двух видов: проверка наблюдений и создание биологических выводов. Из-за ошибок дифференциальной экспрессии и секвенирования РНК важные наблюдения повторяются с помощью (1) ортогонального метода в тех же образцах (например, ПЦР в реальном времени ) или (2) другого, иногда заранее зарегистрированного , эксперимента в новой когорте. . Последнее помогает обеспечить возможность обобщения и обычно может сопровождаться метаанализом всех объединенных когорт. Наиболее распространенным методом получения биологического понимания результатов более высокого уровня является анализ обогащения набора генов , хотя иногда используются подходы с генами-кандидатами. Обогащение набора генов определяет, является ли перекрытие между двумя наборами генов статистически значимым, в данном случае перекрытие между дифференциально экспрессируемыми генами и наборами генов из известных путей/баз данных ( например , Онтология генов , KEGG , Онтология фенотипа человека ) или из дополнительных анализов в одни и те же данные (например, сети совместного выражения). Общие инструменты для обогащения набора генов включают веб-интерфейсы ( например , ENRICHR, g:profiles, WEBGESTALT) [117] и пакеты программного обеспечения. При оценке результатов обогащения одна из эвристик заключается в том, чтобы сначала искать обогащение известной биологии в качестве проверки работоспособности, а затем расширять область поиска для поиска новой биологии.
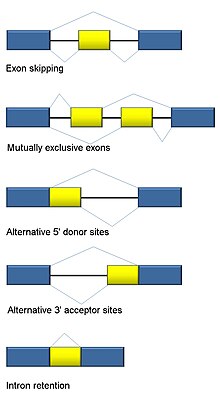
Альтернативный сплайсинг
[ редактировать ]Сплайсинг РНК является неотъемлемой частью эукариот и вносит значительный вклад в регуляцию и разнообразие белков, встречаясь более чем в 90% генов человека. [118] Существует несколько альтернативных режимов сплайсинга : пропуск экзонов (наиболее распространенный режим сплайсинга у людей и высших эукариот), взаимоисключающие экзоны, альтернативные донорные или акцепторные сайты, удержание интрона (наиболее распространенный режим сплайсинга у растений, грибов и простейших), альтернативное начало транскрипции. сайт (промотор) и альтернативное полиаденилирование. [118] Одна из целей RNA-Seq — идентифицировать альтернативные события сплайсинга и проверить, различаются ли они в разных условиях. Секвенирование длинного считывания фиксирует полный транскрипт и, таким образом, сводит к минимуму многие проблемы при оценке численности изоформ, такие как неоднозначное картирование чтения. Для короткого считывания RNA-Seq существует несколько методов обнаружения альтернативного сплайсинга, которые можно разделить на три основные группы: [119] [91] [120]
- На основе подсчета (также на основе событий, дифференциального сплайсинга): оценка удержания экзонов. Примеры: DEXSeq, [121] МАТС, [122] и SeqGSEA. [123]
- На основе изоформ (также модули многократного чтения, дифференциальное выражение изоформ) : сначала оценивают численность изоформ, а затем относительную численность между условиями. Примеры: Запонки 2. [124] и ДиффСплайс. [125]
- На основе вырезания интрона: расчет альтернативного сплайсинга с использованием разделенных чтений. Примеры: MAJIQ [126] и Листорез. [120]
Инструменты дифференциальной экспрессии генов также можно использовать для дифференциальной экспрессии изоформ, если изоформы заранее количественно определены с помощью других инструментов, таких как RSEM. [127]
Сети коэкспрессии
[ редактировать ]Сети коэкспрессии — это полученные на основе данных представления генов, которые ведут себя одинаково в разных тканях и экспериментальных условиях. [128] Их основная цель заключается в создании гипотез и подходах на основе ассоциации вины для вывода о функциях ранее неизвестных генов. [128] Данные RNA-Seq использовались для вывода о генах, участвующих в определенных путях, на основе корреляции Пирсона , как у растений, так и у растений. [129] и млекопитающие. [130] Основным преимуществом данных RNA-Seq в этом виде анализа перед платформами микрочипов является способность охватить весь транскриптом, что дает возможность раскрыть более полные представления о регуляторных сетях генов. Дифференциальную регуляцию сплайсинговых изоформ одного и того же гена можно обнаружить и использовать для прогнозирования их биологических функций. [131] [132] Анализ сети взвешенной совместной экспрессии генов успешно использовался для идентификации модулей совместной экспрессии и внутримодульных генов-концентраторов на основе данных секвенирования РНК. Модули совместной экспрессии могут соответствовать типам клеток или путям. Внутримодульные концентраторы с высокой степенью связи можно интерпретировать как представителей соответствующего модуля. Собственный ген — это взвешенная сумма экспрессии всех генов в модуле. Собственные гены являются полезными биомаркерами (признаками) для диагностики и прогноза. [133] Были предложены подходы к стабилизирующему дисперсию преобразованию для оценки коэффициентов корреляции на основе данных секвенирования РНК. [129]
Открытие вариантов
[ редактировать ]RNA-Seq фиксирует вариации ДНК, включая однонуклеотидные варианты , небольшие вставки/делеции . и структурные вариации . Вызов вариантов в RNA-Seq аналогичен вызову вариантов ДНК и часто использует те же инструменты (включая SAMtools mpileup [134] и GATK HaplotypeCaller [135] ) с поправками для учета сращивания. Одним из уникальных аспектов вариантов РНК является аллель-специфическая экспрессия (ASE) : варианты только одного гаплотипа могут экспрессироваться преимущественно из-за регуляторных эффектов, включая импринтинг и экспрессию локусов количественных признаков , а также некодирующих редких вариантов . [136] [137] Ограничения идентификации вариантов РНК включают то, что она отражает только экспрессированные области (у людей <5% генома) и может быть подвержена систематическим ошибкам, вызванным обработкой данных (например, сборки транскриптома de novo недооценивают гетерозиготность). [138] ), и имеет более низкое качество по сравнению с прямым секвенированием ДНК.
Редактирование РНК (посттранскрипционные изменения)
[ редактировать ]Наличие совпадающих геномных и транскриптомных последовательностей человека может помочь обнаружить посттранскрипционные изменения ( редактирование РНК ). [3] Событие посттранскрипционной модификации идентифицируется, если транскрипт гена имеет аллель/вариант, не наблюдаемый в геномных данных.
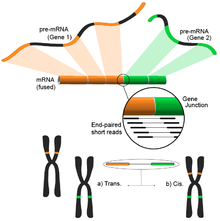
Обнаружение слитых генов
[ редактировать ]Вызванные различными структурными модификациями генома, слитые гены привлекли внимание из-за их связи с раком. [139] Способность RNA-Seq беспристрастно анализировать весь транскриптом образца делает его привлекательным инструментом для выявления подобных распространенных явлений при раке. [4]
Идея вытекает из процесса сопоставления коротких транскриптомных чтений с эталонным геномом. Большинство коротких ридов будут находиться в пределах одного полного экзона, а меньший, но все же большой набор, как ожидается, будет картироваться на известных соединениях экзон-экзон. Оставшиеся некартированные короткие чтения затем будут дополнительно проанализированы, чтобы определить, соответствуют ли они соединению экзон-экзон, где экзоны происходят из разных генов. Это было бы свидетельством возможного события слияния, однако из-за продолжительности считываний это может оказаться очень шумным. Альтернативный подход заключается в использовании парных чтений, когда потенциально большое количество парных ридов будет сопоставлять каждый конец с различным экзоном, обеспечивая лучшее освещение этих событий (см. рисунок). Тем не менее, конечный результат состоит из множества потенциально новых комбинаций генов, обеспечивающих идеальную отправную точку для дальнейшей проверки.
Копирование изменения номера
[ редактировать ]Анализ изменения числа копий (CNA) обычно используется в исследованиях рака. Приобретение и потеря генов имеют значение для сигнального пути и являются ключевым биомаркером молекулярной дисфункции в онкологии. Вызвать информацию CNA из данных RNA-Seq непросто из-за различий в экспрессии генов, которые приводят к разной величине различий в глубине считывания между генами. Из-за этих трудностей большая часть этих анализов обычно проводится с использованием полногеномного секвенирования/секвенирования всего экзома (WGS/WES). Но передовые инструменты биоинформатики могут вызывать CNA из RNA-Seq. [140]
Другие новые методы анализа и применения
[ редактировать ]Области применения RNA-Seq растут с каждым днем. Другое новое применение RNA-Seq включает обнаружение микробных примесей, [141] определение численности типов клеток (деконволюция типов клеток), [8] измерение экспрессии TE и предсказание неоантигена и т. д. [8]
История
[ редактировать ]
RNA-Seq был впервые разработан в середине 2000-х годов с появлением технологии секвенирования нового поколения. [144] Первые рукописи, в которых использовалась RNA-Seq даже без использования этого термина, включают в себя исследования рака простаты. клеточных линий [145] (от 2006 г.), Medicago truncatula [146] (2006), кукуруза [147] (2007) и Arabidopsis thaliana [148] (2007), а сам термин «RNA-Seq» впервые был упомянут в 2008 году. [13] [149] Количество рукописей, упоминающих RNA-Seq в названии или аннотации (рисунок, синяя линия), постоянно увеличивается: в 2018 году было опубликовано 6754 рукописи. Пересечение RNA-Seq и медицины (рисунок, золотая линия) происходит с такой же скоростью. [150]
Приложения в медицине
[ редактировать ]RNA-Seq имеет потенциал для выявления новой биологии заболеваний, составления профиля биомаркеров для клинических показаний, определения путей, подходящих для лекарств, и постановки генетических диагнозов. [151] [152] Эти результаты могут быть дополнительно персонализированы для подгрупп или даже отдельных пациентов, потенциально подчеркивая более эффективную профилактику, диагностику и терапию. Осуществимость этого подхода частично продиктована затратами денег и времени; связанным ограничением является необходимость в команде специалистов (биоинформатиков, врачей/клиницистов, фундаментальных исследователей, технических специалистов) для полной интерпретации огромного количества данных, полученных в результате этого анализа. [153]
Масштабные усилия по секвенированию
[ редактировать ]Большое внимание было уделено данным RNA-Seq после того, как проекты «Энциклопедия элементов ДНК» (ENCODE) и «Атлас генома рака» (TCGA) использовали этот подход для характеристики десятков клеточных линий. [154] и тысячи образцов первичных опухолей, [155] соответственно. ENCODE нацелена на идентификацию полногеномных регуляторных областей в различных группах клеточных линий, и транскриптомные данные имеют первостепенное значение для понимания последующего эффекта этих эпигенетических и генетических регуляторных слоев. Вместо этого TCGA стремилась собрать и проанализировать тысячи образцов пациентов из 30 различных типов опухолей, чтобы понять основные механизмы злокачественной трансформации и прогрессирования. В этом контексте данные RNA-Seq дают уникальную картину транскриптомного статуса заболевания и рассматривают беспристрастную популяцию транскриптов, что позволяет идентифицировать новые транскрипты, слитые транскрипты и некодирующие РНК, которые можно было бы не обнаружить с помощью различных технологий.
См. также
[ редактировать ]Ссылки
[ редактировать ]![]() Эта статья была отправлена в WikiJournal of Science на внешнюю академическую рецензию в 2019 году ( отчеты рецензентов ). Обновленный контент был реинтегрирован на страницу Википедии по лицензии CC-BY-SA-3.0 ( 2021 г. ). Проверенная версия записи: Феликс Рихтер и др. (17 мая 2021 г.). «Общее введение в секвенирование РНК» (PDF) . Викижурнал науки 4 (2):4.doi : 10.15347 /WJS/2021.004 . ISSN 2470-6345 . Викиданные Q100146647 .
Эта статья была отправлена в WikiJournal of Science на внешнюю академическую рецензию в 2019 году ( отчеты рецензентов ). Обновленный контент был реинтегрирован на страницу Википедии по лицензии CC-BY-SA-3.0 ( 2021 г. ). Проверенная версия записи: Феликс Рихтер и др. (17 мая 2021 г.). «Общее введение в секвенирование РНК» (PDF) . Викижурнал науки 4 (2):4.doi : 10.15347 /WJS/2021.004 . ISSN 2470-6345 . Викиданные Q100146647 .
- ^ Лоу Р., Ширли Н., Бликли М., Долан С., Шафи Т. (май 2017 г.). «Технологии транскриптомики» . PLOS Вычислительная биология . 13 (5): e1005457. Бибкод : 2017PLSCB..13E5457L . дои : 10.1371/journal.pcbi.1005457 . ПМЦ 5436640 . ПМИД 28545146 .
- ^ Чу Ю, Кори Д.Р. (август 2012 г.). «Секвенирование РНК: выбор платформы, дизайн эксперимента и интерпретация данных» . Нуклеиновая кислотная терапия . 22 (4): 271–4. дои : 10.1089/нат.2012.0367 . ПМК 3426205 . ПМИД 22830413 .
- ^ Jump up to: а б с Ван З., Герштейн М., Снайдер М. (январь 2009 г.). «RNA-Seq: революционный инструмент для транскриптомики» . Обзоры природы. Генетика . 10 (1): 57–63. дои : 10.1038/nrg2484 . ПМЦ 2949280 . ПМИД 19015660 .
- ^ Jump up to: а б Махер К.А., Кумар-Синха С., Цао Х, Кальяна-Сундарам С., Хан Б., Цзин Х и др. (март 2009 г.). «Секвенирование транскриптома для обнаружения слияний генов при раке» . Природа . 458 (7234): 97–101. Бибкод : 2009Natur.458...97M . дои : 10.1038/nature07638 . ПМК 2725402 . ПМИД 19136943 .
- ^ Инголия НТ, Брар Г.А., Рускин С., Макгичи А.М., Вайсман Дж.С. (июль 2012 г.). «Стратегия профилирования рибосом для мониторинга трансляции in vivo путем глубокого секвенирования фрагментов мРНК, защищенных рибосомами» . Протоколы природы . 7 (8): 1534–50. дои : 10.1038/nprot.2012.086 . ПМК 3535016 . ПМИД 22836135 .
- ^ Альперн Д., Гардо В., Рассел Дж., Манжеат Б., Мейрелеш-Фильо А.С., Брейсс Р. и др. (апрель 2019 г.). «BRB-seq: сверхдоступная высокопроизводительная транскриптомика, обеспечиваемая массовым штрих-кодированием и секвенированием РНК» . Геномная биология . 20 (1): 71. дои : 10.1186/s13059-019-1671-x . ПМК 6474054 . ПМИД 30999927 .
- ^ Ли Дж.Х., Догарти Э.Р., Шейман Дж., Калхор Р., Ян Дж.Л., Ферранте Т.С. и др. (март 2014 г.). «Высокомультиплексное секвенирование субклеточной РНК in situ» . Наука . 343 (6177): 1360–3. Бибкод : 2014Sci...343.1360L . дои : 10.1126/science.1250212 . ПМК 4140943 . ПМИД 24578530 .
- ^ Jump up to: а б с Тинд А.С., Монга И., Тхакур П.К., Кумари П., Диндория К., Крзак М. и др. (ноябрь 2021 г.). «Демистификация новых приложений массового секвенирования РНК: применение и полезность биоинформатической методологии». Брифинги по биоинформатике . 22 (6). дои : 10.1093/нагрудник/bbab259 . ПМИД 34329375 .
- ^ Кукурба К.Р., Монтгомери С.Б. (апрель 2015 г.). «Секвенирование и анализ РНК» . Протоколы Колд-Спринг-Харбора . 2015 (11): 951–69. дои : 10.1101/pdb.top084970 . ПМЦ 4863231 . ПМИД 25870306 .
- ^ Jump up to: а б с д и Гриффит М., Уокер-младший, Spies NC, Эйнскоу Б.Дж., Гриффит О.Л. (август 2015 г.). «Информатика для секвенирования РНК: веб-ресурс для анализа в облаке» . PLOS Вычислительная биология . 11 (8): e1004393. Бибкод : 2015PLSCB..11E4393G . дои : 10.1371/journal.pcbi.1004393 . ПМЦ 4527835 . ПМИД 26248053 .
- ^ «РНК-секлопедия» . rnaseq.uoregon.edu . Проверено 8 февраля 2017 г.
- ^ Морин Р., Бейнбридж М., Фейес А., Херст М., Кшивински М., Пью Т. и др. (июль 2008 г.). «Профилирование транскриптома HeLa S3 с использованием случайно выбранной кДНК и массово-параллельного секвенирования с коротким считыванием» . БиоТехники . 45 (1): 81–94. дои : 10.2144/000112900 . ПМИД 18611170 .
- ^ Jump up to: а б с д Мортазави А., Уильямс Б.А., МакКью К., Шеффер Л., Уолд Б. (июль 2008 г.). «Картирование и количественная оценка транскриптомов млекопитающих с помощью RNA-Seq». Природные методы . 5 (7): 621–8. дои : 10.1038/nmeth.1226 . ПМИД 18516045 . S2CID 205418589 .
- ^ Сунь Ц, Хао Ц, Прасант К.В. (февраль 2018 г.). «Ядерные длинные некодирующие РНК: ключевые регуляторы экспрессии генов» . Тенденции в генетике . 34 (2): 142–157. дои : 10.1016/j.tig.2017.11.005 . ПМК 6002860 . ПМИД 29249332 .
- ^ Сигургерссон Б., Эмануэльссон О., Лундеберг Дж. (2014). «Секвенирование деградированной РНК, адресованной путем подсчета 3'-тегов» . ПЛОС ОДИН . 9 (3): е91851. Бибкод : 2014PLoSO...991851S . дои : 10.1371/journal.pone.0091851 . ПМЦ 3954844 . ПМИД 24632678 .
- ^ Чен Е.А., Суайая Т., Херштейн Дж.С., Евграфов О.В., Спицына В.Н., Реболини Д.Ф. и др. (октябрь 2014 г.). «Влияние целостности РНК на уникально картированные чтения в RNA-Seq» . Исследовательские заметки BMC . 7 : 753. doi : 10.1186/1756-0500-7-753 . ПМЦ 4213542 . ПМИД 25339126 .
- ^ Молл П., Анте М., Зейтц А., Реда Т. (декабрь 2014 г.). «Секвенирование мРНК QuantSeq 3' для количественного определения РНК». Природные методы . 11 (12): i – iii. дои : 10.1038/nmeth.f.376 . ISSN 1548-7105 . S2CID 83424788 .
- ^ Ойкономопулос С., Байега А., Фахиминия С., Джамбазиан Х., Берубе П., Рагуссис Дж. (2020). «Методики профилирования транскриптов с использованием давно известных технологий» . Границы генетики . 11 : 606. дои : 10.3389/fgene.2020.00606 . ПМЦ 7358353 . ПМИД 32733532 .
- ^ Jump up to: а б Конеса А., Мадригал П., Таразона С., Гомес-Кабреро Д., Сервера А., Макферсон А. и др. (январь 2016 г.). «Обзор лучших практик анализа данных секвенирования РНК» . Геномная биология . 17 (1): 13. дои : 10.1186/s13059-016-0881-8 . ПМЦ 4728800 . ПМИД 26813401 .
- ^ Лю Д., Грабер Дж. Х. (февраль 2006 г.). «Количественное сравнение библиотек EST требует компенсации систематических ошибок в генерации кДНК» . БМК Биоинформатика . 7:77 . дои : 10.1186/1471-2105-7-77 . ПМЦ 1431573 . ПМИД 16503995 .
- ^ Jump up to: а б с Гаральде Д.Р., Снелл Э.А., Яхимович Д., Сипос Б., Ллойд Дж.Х., Брюс М. и др. (март 2018 г.). «Высокопараллельное прямое секвенирование РНК на массиве нанопор». Природные методы . 15 (3): 201–206. дои : 10.1038/nmeth.4577 . ПМИД 29334379 . S2CID 3589823 .
- ^ Лю Д., Грабер Дж. Х. (февраль 2006 г.). «Количественное сравнение библиотек EST требует компенсации систематических ошибок в генерации кДНК» . БМК Биоинформатика . 7:77 . дои : 10.1186/1471-2105-7-77 . ПМЦ 1431573 . ПМИД 16503995 .
- ^ Глисон Дж., Лейн Т.А., Харрисон П.Дж., Харти В., Кларк М.Б. (3 августа 2020 г.). «Прямое секвенирование РНК нанопорами обнаруживает дифференциальную экспрессию между популяциями клеток человека» . bioRxiv : 2020.08.02.232785. дои : 10.1101/2020.08.02.232785 . S2CID 220975367 .
- ^ Jump up to: а б " Шапиро Э., Бизунер Т., Линнарссон С. (сентябрь 2013 г.). «Технологии, основанные на секвенировании отдельных клеток, произведут революцию в науке о целом организме». Обзоры природы. Генетика . 14 (9): 618–30. дои : 10.1038/nrg3542 . ПМИД 23897237 . S2CID 500845 . "
- ^ Колодзейчик А.А., Ким Дж.К., Свенссон В., Мариони Дж.К., Тейхманн С.А. (май 2015 г.). «Технология и биология секвенирования одноклеточных РНК» . Молекулярная клетка . 58 (4): 610–20. doi : 10.1016/j.molcel.2015.04.005 . ПМИД 26000846 .
- ^ Монторо Д.Т., Хабер А.Л., Битон М., Винарский В., Лин Б., Биркет С.Е. и др. (август 2018 г.). «Пересмотренная иерархия эпителия дыхательных путей включает ионоциты, экспрессирующие CFTR» . Природа . 560 (7718): 319–324. Бибкод : 2018Natur.560..319M . дои : 10.1038/s41586-018-0393-7 . ПМК 6295155 . ПМИД 30069044 .
- ^ Пласшерт Л.В., Жилионис Р., Чу-Винг Р., Савова В., Кнер Дж., Рома Г. и др. (август 2018 г.). «Одноклеточный атлас эпителия дыхательных путей обнаруживает богатые CFTR легочные ионоциты» . Природа . 560 (7718): 377–381. Бибкод : 2018Natur.560..377P . дои : 10.1038/s41586-018-0394-6 . ПМК 6108322 . ПМИД 30069046 .
- ^ Валиграч Л., Андрович П., Кубиста М. (март 2018 г.). «Платформы для сбора и анализа отдельных клеток» . Международный журнал молекулярных наук . 19 (3): 807. doi : 10.3390/ijms19030807 . ПМК 5877668 . ПМИД 29534489 .
- ^ Кляйн А.М., Мазутис Л., Акартуна И., Таллапрагада Н., Верес А., Ли В. и др. (май 2015 г.). «Капельное штрих-кодирование для транскриптомики отдельных клеток применительно к эмбриональным стволовым клеткам» . Клетка . 161 (5): 1187–1201. дои : 10.1016/j.cell.2015.04.044 . ПМЦ 4441768 . ПМИД 26000487 .
- ^ Макоско Э.З., Басу А., Сатия Р., Немеш Дж., Шекхар К., Голдман М. и др. (май 2015 г.). «Высокопараллельное полногеномное профилирование экспрессии отдельных клеток с использованием нанолитровых капель» . Клетка . 161 (5): 1202–1214. дои : 10.1016/j.cell.2015.05.002 . ПМЦ 4481139 . ПМИД 26000488 .
- ^ Ислам С., Зейзель А., Йост С., Ла Манно Г., Заяк П., Каспер М. и др. (февраль 2014 г.). «Количественный секвенирование одноклеточной РНК с уникальными молекулярными идентификаторами». Природные методы . 11 (2): 163–6. дои : 10.1038/nmeth.2772 . ПМИД 24363023 . S2CID 6765530 .
- ^ " Хебенстрейт Д. (ноябрь 2012 г.). «Методы, проблемы и возможности секвенирования одноклеточной РНК» . Биология . 1 (3): 658–67. дои : 10.3390/biology1030658 . ПМК 4009822 . ПМИД 24832513 . "
- ^ Эбервин Дж., Сул Дж.Й., Бартфай Т., Ким Дж. (январь 2014 г.). «Обещание секвенирования отдельных клеток». Природные методы . 11 (1): 25–7. дои : 10.1038/nmeth.2769 . ПМИД 24524134 . S2CID 11575439 .
- ^ Тан Ф., Барбачору С., Ван Ю., Нордман Э., Ли С., Сюй Н. и др. (май 2009 г.). «Анализ всего транскриптома мРНК-Seq одной клетки». Природные методы . 6 (5): 377–82. дои : 10.1038/NMETH.1315 . ПМИД 19349980 . S2CID 16570747 .
- ^ Ислам С., Кьеллквист У., Молинер А., Заяц П., Фан Дж.Б., Лённерберг П. и др. (июль 2011 г.). «Характеристика одноклеточного транскрипционного ландшафта с помощью высокомультиплексного секвенирования РНК» . Геномные исследования . 21 (7): 1160–7. дои : 10.1101/гр.110882.110 . ПМЦ 3129258 . ПМИД 21543516 .
- ^ Рамшельд Д., Луо С., Ван Ю.К., Ли Р., Дэн К., Фаридани О.Р. и др. (август 2012 г.). «Полноразмерная мРНК-Seq из одноклеточных уровней РНК и отдельных циркулирующих опухолевых клеток» . Природная биотехнология . 30 (8): 777–82. дои : 10.1038/nbt.2282 . ПМЦ 3467340 . ПМИД 22820318 .
- ^ Хашимшони Т., Вагнер Ф., Шер Н., Янаи И. (сентябрь 2012 г.). «CEL-Seq: Seq одноклеточной РНК путем мультиплексной линейной амплификации» . Отчеты по ячейкам . 2 (3): 666–73. дои : 10.1016/j.celrep.2012.08.003 . ПМИД 22939981 .
- ^ Сингх М., Аль-Эриани Г., Карсвелл С., Фергюсон Дж. М., Блэкберн Дж., Бартон К. и др. (2018). «Высокопроизводительное целевое секвенирование отдельных клеток с длинным считыванием раскрывает клональный и транскрипционный ландшафт лимфоцитов» . биоRxiv . 10 (1): 3120. дои : 10.1101/424945 . ПМЦ 6635368 . ПМИД 31311926 .
- ^ Сасагава Ю., Никайдо И., Хаяси Т., Данно Х., Уно К.Д., Имаи Т. и др. (апрель 2013 г.). «Quartz-Seq: высоковоспроизводимый и чувствительный метод секвенирования одноклеточной РНК, выявляющий негенетическую гетерогенность экспрессии генов» . Геномная биология . 14 (4): С31. дои : 10.1186/gb-2013-14-4-r31 . ПМК 4054835 . ПМИД 23594475 .
- ^ Коуно Т., Муди Дж., Квон А.Т., Сибаяма Ю., Като С., Хуан Ю. и др. (январь 2019 г.). «C1 CAGE обнаруживает сайты начала транскрипции и активность энхансера с разрешением одной клетки» . Природные коммуникации . 10 (1): 360. Бибкод : 2019NatCo..10..360K . дои : 10.1038/s41467-018-08126-5 . ПМК 6341120 . ПМИД 30664627 .
- ^ Даль Молин А, Ди Камилло Б (2019). «Как разработать эксперимент по секвенированию одноклеточной РНК: подводные камни, проблемы и перспективы». Брифинги по биоинформатике . 20 (4): 1384–1394. дои : 10.1093/нагрудник/bby007 . ПМИД 29394315 .
- ^ Петерсон В.М., Чжан К.Х., Кумар Н., Вонг Дж., Ли Л., Уилсон Д.С. и др. (октябрь 2017 г.). «Мультиплексная количественная оценка белков и транскриптов в отдельных клетках». Природная биотехнология . 35 (10): 936–939. дои : 10.1038/nbt.3973 . ПМИД 28854175 . S2CID 205285357 .
- ^ Стоккиус М., Хафемейстер С., Стивенсон В., Хоук-Лумис Б., Чаттопадхьяй П.К., Свердлов Х. и др. (сентябрь 2017 г.). «Одновременное измерение эпитопа и транскриптома в отдельных клетках» . Природные методы . 14 (9): 865–868. дои : 10.1038/nmeth.4380 . ПМК 5669064 . ПМИД 28759029 .
- ^ Радж Б., Вагнер Д.Э., Маккенна А., Панди С., Кляйн А.М., Шендюр Дж. и др. (июнь 2018 г.). «Одновременное одноклеточное профилирование линий и типов клеток в мозге позвоночных» . Природная биотехнология . 36 (5): 442–450. дои : 10.1038/nbt.4103 . ПМЦ 5938111 . ПМИД 29608178 .
- ^ Олмос Д., Аркенау Х.Т., Анг Дж.Е., Ледаки И., Аттард Г., Карден К.П. и др. (январь 2009 г.). «Циркулирующие опухолевые клетки (ЦОК) считаются промежуточными конечными точками при кастрационно-резистентном раке простаты (КРРПЖ): опыт одного центра» . Анналы онкологии . 20 (1): 27–33. дои : 10.1093/annonc/mdn544 . ПМИД 18695026 .
- ^ Левитин Х.М., Юань Дж., Симс П.А. (апрель 2018 г.). «Одноклеточный транскриптомный анализ гетерогенности опухоли» . Тенденции рака . 4 (4): 264–268. дои : 10.1016/j.trecan.2018.02.003 . ПМЦ 5993208 . ПМИД 29606308 .
- ^ Джерби-Арнон Л., Шах П., Куоко М.С., Родман С., Су М.Дж., Мелмс Дж.К. и др. (ноябрь 2018 г.). «Программа по борьбе с раковыми клетками способствует исключению Т-клеток и устойчивости к блокаде контрольных точек» . Клетка . 175 (4): 984–997.e24. дои : 10.1016/j.cell.2018.09.006 . ПМК 6410377 . ПМИД 30388455 .
- ^ Стивенсон В., Донлин Л.Т., Батлер А., Розо С., Бракен Б., Рашидфаррохи А. и др. (февраль 2018 г.). «Секвенирование одноклеточной РНК синовиальной ткани ревматоидного артрита с использованием недорогих микрофлюидных инструментов» . Природные коммуникации . 9 (1): 791. Бибкод : 2018NatCo...9..791S . дои : 10.1038/s41467-017-02659-x . ПМЦ 5824814 . ПМИД 29476078 .
- ^ Авраам Р., Хасли Н., Браун Д., Пенаранда С., Джиджон Х.Б., Тромбетта Дж.Дж. и др. (сентябрь 2015 г.). «Межклеточная изменчивость патогена приводит к гетерогенности иммунных реакций хозяина» . Клетка . 162 (6): 1309–21. дои : 10.1016/j.cell.2015.08.027 . ПМЦ 4578813 . ПМИД 26343579 .
- ^ Цао Дж., Пакер Дж.С., Рамани В., Кусанович Д.А., Хуинь С., Даза Р. и др. (август 2017 г.). «Комплексное одноклеточное транскрипционное профилирование многоклеточного организма» . Наука . 357 (6352): 661–667. Бибкод : 2017Sci...357..661C . дои : 10.1126/science.aam8940 . ПМЦ 5894354 . ПМИД 28818938 .
- ^ Пласс М., Солана Дж., Вольф Ф.А., Аюб С., Мисиос А., Глазар П. и др. (май 2018 г.). «Атлас типов клеток и древо происхождения целого сложного животного по данным одноклеточной транскриптомики» . Наука . 360 (6391): eaaq1723. дои : 10.1126/science.aaq1723 . ПМИД 29674432 .
- ^ Финчер К.Т., Вурцель О., де Хоог Т., Краварик К.М., Реддиен П.В. (май 2018 г.). «Средиземноморская Шмидтея » Наука 360 (6391): eaaq1 дои : 10.1126/science.aaq1736 . ПМК 6563842 . ПМИД 29674431 .
- ^ Вагнер Д.Э., Вайнреб С., Коллинз З.М., Бриггс Дж.А., Мегасон С.Г., Кляйн А.М. (июнь 2018 г.). «Одноклеточное картирование ландшафтов экспрессии генов и их происхождения у эмбрионов рыбок данио» . Наука . 360 (6392): 981–987. Бибкод : 2018Sci...360..981W . дои : 10.1126/science.aar4362 . ПМК 6083445 . ПМИД 29700229 .
- ^ Фаррелл Дж.А., Ван Й., Ризенфельд С.Дж., Шекхар К., Регев А., Шир А.Ф. (июнь 2018 г.). «Одноклеточная реконструкция траекторий развития во время эмбриогенеза рыбок данио» . Наука . 360 (6392): eaar3131. дои : 10.1126/science.aar3131 . ПМК 6247916 . ПМИД 29700225 .
- ^ Бриггс Дж.А., Вайнреб С., Вагнер Д.Е., Мегасон С., Пешкин Л., Киршнер М.В. и др. (июнь 2018 г.). «Динамика экспрессии генов в эмбриогенезе позвоночных при разрешении отдельных клеток» . Наука . 360 (6392): eaar5780. дои : 10.1126/science.aar5780 . ПМК 6038144 . ПМИД 29700227 .
- ^ Ю Дж. «Научный прорыв 2018 года: отслеживание развития клетка за клеткой» . Научный журнал . Американская ассоциация содействия развитию науки.
- ^ Jump up to: а б Ли Х, Ловчи М.Т., Квон Ю.С., Розенфельд М.Г., Фу XD, Йео Г.В. (декабрь 2008 г.). «Определение плотности меток, необходимой для анализа цифрового транскриптома: применение к андроген-чувствительной модели рака простаты» . Труды Национальной академии наук Соединенных Штатов Америки . 105 (51): 20179–84. Бибкод : 2008PNAS..10520179L . дои : 10.1073/pnas.0807121105 . ПМК 2603435 . ПМИД 19088194 .
- ^ Jump up to: а б Стегл О., Партс Л., Пийпари М., Винн Дж., Дурбин Р. (февраль 2012 г.). «Использование вероятностной оценки остатков экспрессии (PEER) для повышения мощности и интерпретируемости анализа экспрессии генов» . Протоколы природы . 7 (3): 500–7. дои : 10.1038/nprot.2011.457 . ПМЦ 3398141 . ПМИД 22343431 .
- ^ Кингсфорд С., Патро Р. (июнь 2015 г.). «Основное сжатие коротких последовательностей чтения с использованием кодирования пути» . Биоинформатика . 31 (12): 1920–8. doi : 10.1093/биоинформатика/btv071 . ПМЦ 4481695 . ПМИД 25649622 .
- ^ Jump up to: а б Грабхерр М.Г., Хаас Б.Дж., Яссур М., Левин Дж.З., Томпсон Д.А., Амит И. и др. (май 2011 г.). «Сборка полноразмерного транскриптома на основе данных RNA-Seq без эталонного генома» . Природная биотехнология . 29 (7): 644–52. дои : 10.1038/nbt.1883 . ПМЦ 3571712 . ПМИД 21572440 .
- ^ «Сборка De Novo с использованием чтения Illumina» (PDF) . Проверено 22 октября 2016 г. .
- ^ Оазисы: ассемблер транскриптома для очень коротких чтений.
- ^ Зербино Д.Р., Бирни Э. (май 2008 г.). «Бархат: алгоритмы сборки короткого чтения de novo с использованием графов де Брёйна» . Геномные исследования . 18 (5): 821–9. дои : 10.1101/гр.074492.107 . ПМК 2336801 . ПМИД 18349386 .
- ^ Чанг З., Ли Г., Лю Дж., Чжан Ю., Эшби С., Лю Д. и др. (февраль 2015 г.). «Bridger: новая основа для сборки транскриптома de novo с использованием данных RNA-seq» . Геномная биология . 16 (1): 30. дои : 10.1186/s13059-015-0596-2 . ПМЦ 4342890 . ПМИД 25723335 .
- ^ Бушманова Е, Антипов Д, Лапидус А, Пржибельски А.Д. (сентябрь 2019 г.). «rnaSPAdes: ассемблер транскриптома de novo и его применение к данным RNA-Seq» . ГигаСайенс . 8 (9). doi : 10.1093/gigascience/giz100 . ПМК 6736328 . ПМИД 31494669 .
- ^ Jump up to: а б Ли Б., Филлмор Н., Бай Ю., Коллинз М., Томсон Дж.А., Стюарт Р. и др. (декабрь 2014 г.). «Оценка сборок транскриптома de novo на основе данных RNA-Seq» . Геномная биология . 15 (12): 553. дои : 10.1186/s13059-014-0553-5 . ПМК 4298084 . ПМИД 25608678 .
- ^ Jump up to: а б Добин А., Дэвис К.А., Шлезингер Ф., Дренков Дж., Залески С., Джа С. и др. (январь 2013 г.). «STAR: сверхбыстрый универсальный выравниватель RNA-seq» . Биоинформатика . 29 (1): 15–21. doi : 10.1093/биоинформатика/bts635 . ПМК 3530905 . ПМИД 23104886 .
- ^ Лэнгмид Б. , Трапнелл С., Поп М., Зальцберг С.Л. (2009). «Сверхбыстрое и эффективное для памяти выравнивание коротких последовательностей ДНК с геномом человека» . Геномная биология . 10 (3): 25 рандов. дои : 10.1186/gb-2009-10-3-r25 . ПМК 2690996 . ПМИД 19261174 .
- ^ Трапнелл С., Пахтер Л. , Зальцберг С.Л. (май 2009 г.). «TopHat: обнаружение соединений сплайсинга с помощью RNA-Seq» . Биоинформатика . 25 (9): 1105–11. doi : 10.1093/биоинформатика/btp120 . ПМЦ 2672628 . ПМИД 19289445 .
- ^ Jump up to: а б Трапнелл С., Робертс А., Гофф Л., Пертеа Г., Ким Д., Келли Д.Р. и др. (март 2012 г.). «Дифференциальный анализ экспрессии генов и транскриптов в экспериментах по секвенированию РНК с помощью TopHat и Cufflinks» . Протоколы природы . 7 (3): 562–78. дои : 10.1038/нпрот.2012.016 . ПМЦ 3334321 . ПМИД 22383036 .
- ^ Ляо Ю., Смит Г.К., Ши В. (май 2013 г.). «Выравниватель Subread: быстрое, точное и масштабируемое сопоставление чтения путем посева и голосования» . Исследования нуклеиновых кислот . 41 (10): е108. дои : 10.1093/нар/gkt214 . ПМЦ 3664803 . ПМИД 23558742 .
- ^ Ким Д., Лэнгмид Б., Зальцберг С.Л. (апрель 2015 г.). «HISAT: выравниватель для быстрого сращивания с низкими требованиями к памяти» . Природные методы . 12 (4): 357–60. дои : 10.1038/nmeth.3317 . ПМЦ 4655817 . ПМИД 25751142 .
- ^ Ву Т.Д., Ватанабэ С.К. (май 2005 г.). «GMAP: программа геномного картирования и выравнивания последовательностей мРНК и EST» . Биоинформатика . 21 (9): 1859–75. doi : 10.1093/биоинформатика/bti310 . ПМИД 15728110 .
- ^ Пертеа М., Пертеа ГМ, Антонеску КМ, Чанг ТК, Менделл Дж.Т., Зальцберг С.Л. (март 2015 г.). «StringTie позволяет улучшить реконструкцию транскриптома на основе считываний RNA-seq» . Природная биотехнология . 33 (3): 290–5. дои : 10.1038/nbt.3122 . ПМЦ 4643835 . ПМИД 25690850 .
- ^ Баруццо Дж., Хайер К.Е., Ким Э.Дж., Ди Камилло Б., Фитцджеральд Г.А., Грант Г.Р. (февраль 2017 г.). «Комплексный сравнительный анализ элайнеров RNA-seq на основе моделирования» . Природные методы . 14 (2): 135–139. дои : 10.1038/nmeth.4106 . ПМК 5792058 . ПМИД 27941783 .
- ^ Энгстрем П.Г., Стейгер Т., Сипос Б., Грант Г.Р., Калес А., Ретш Г. и др. (декабрь 2013 г.). «Систематическая оценка программ выравнивания сплайсинга для данных секвенирования РНК» . Природные методы . 10 (12): 1185–91. дои : 10.1038/nmeth.2722 . ПМК 4018468 . ПМИД 24185836 .
- ^ Лу Б, Цзэн Цзи, Ши Т (февраль 2013 г.). «Сравнительное исследование стратегий сборки de novo и сборки на основе генома для реконструкции транскриптома на основе RNA-Seq» . Наука Китай Науки о жизни . 56 (2): 143–55. дои : 10.1007/s11427-013-4442-z . ПМИД 23393030 .
- ^ Брэднам К.Р., Фасс Дж.Н., Александров А., Баранай П., Бехнер М., Бироль И. и др. (июль 2013 г.). «Сборка 2: оценка методов сборки генома de novo у трех видов позвоночных» . ГигаСайенс . 2 (1): 10. arXiv : 1301.5406 . Бибкод : 2013arXiv1301.5406B . дои : 10.1186/2047-217X-2-10 . ПМЦ 3844414 . ПМИД 23870653 .
- ^ Хельцер М., Марц М. (май 2019 г.). «Сборка транскриптома de novo: всестороннее межвидовое сравнение ассемблеров короткого чтения RNA-Seq» . ГигаСайенс . 8 (5). doi : 10.1093/gigascience/giz039 . ПМК 6511074 . ПМИД 31077315 .
- ^ Гринбаум Д., Коланджело С., Уильямс К., Герштейн М. (2003). «Сравнение количества белков и уровней экспрессии мРНК в геномном масштабе» . Геномная биология . 4 (9): 117. doi : 10.1186/gb-2003-4-9-117 . ЧВК 193646 . ПМИД 12952525 .
- ^ Чжан З.Х., Джавери Д.Д., Маршалл В.М., Бауэр Д.С., Эдсон Дж., Нарайанан Р.К. и др. (август 2014 г.). «Сравнительное исследование методов дифференциального анализа экспрессии данных RNA-Seq» . ПЛОС ОДИН . 9 (8): е103207. Бибкод : 2014PLoSO...9j3207Z . дои : 10.1371/journal.pone.0103207 . ПМК 4132098 . ПМИД 25119138 .
- ^ Андерс С., Пил П.Т., Хубер В. (январь 2015 г.). «HTSeq — платформа Python для работы с данными высокопроизводительного секвенирования» . Биоинформатика . 31 (2): 166–9. doi : 10.1093/биоинформатика/btu638 . ПМК 4287950 . ПМИД 25260700 .
- ^ Ляо Ю., Смит Г.К., Ши В. (апрель 2014 г.). «featureCounts: эффективная программа общего назначения для присвоения считывания последовательностей геномным признакам». Биоинформатика . 30 (7): 923–30. arXiv : 1305.3347 . doi : 10.1093/биоинформатика/btt656 . ПМИД 24227677 .
- ^ Шмид М.В., Гроссниклаус У (февраль 2015 г.). «Rcount: простой и гибкий подсчет чтений RNA-Seq» . Биоинформатика . 31 (3): 436–7. doi : 10.1093/биоинформатика/btu680 . ПМИД 25322836 .
- ^ Финотелло Ф., Лавеццо Э., Бьянко Л., Барзон Л., Маццон П., Фонтана П. и др. (2014). «Уменьшение систематической ошибки в данных секвенирования РНК: новый подход к подсчету количества» . БМК Биоинформатика . 15 (Приложение 1): S7. дои : 10.1186/1471-2105-15-s1-s7 . ПМК 4016203 . ПМИД 24564404 .
- ^ Хасимото ТБ, Эдвардс, доктор медицины, Гиффорд Д.К. (март 2014 г.). «Универсальная коррекция счета для высокопроизводительного секвенирования» . PLOS Вычислительная биология . 10 (3): e1003494. Бибкод : 2014PLSCB..10E3494H . дои : 10.1371/journal.pcbi.1003494 . ПМЦ 3945112 . ПМИД 24603409 .
- ^ Патро Р., Маунт СМ, Кингсфорд К. (май 2014 г.). «Sailfish позволяет проводить количественный анализ изоформ без выравнивания на основе считываний РНК-seq с использованием облегченных алгоритмов» . Природная биотехнология . 32 (5): 462–4. arXiv : 1308.3700 . дои : 10.1038/nbt.2862 . ПМК 4077321 . ПМИД 24752080 .
- ^ Брэй Н.Л., Пиментел Х., Мелстед П., Пахтер Л. (май 2016 г.). «Почти оптимальная вероятностная количественная оценка секвенирования РНК» . Природная биотехнология . 34 (5): 525–7. дои : 10.1038/nbt.3519 . ПМИД 27043002 . S2CID 205282743 .
- ^ Jump up to: а б Робинсон, доктор медицинских наук, Ошлак А. (2010). «Метод нормализации масштабирования для анализа дифференциальной экспрессии данных секвенирования РНК» . Геномная биология . 11 (3): 25 р. дои : 10.1186/gb-2010-11-3-r25 . ПМЦ 2864565 . ПМИД 20196867 .
- ^ Трапнелл С., Уильямс Б.А., Пертеа Г., Мортазави А., Кван Г., ван Барен М.Дж. и др. (май 2010 г.). «Сборка транскриптов и количественная оценка с помощью RNA-Seq выявляют неаннотированные транскрипты и переключение изоформ во время дифференцировки клеток» . Природная биотехнология . 28 (5): 511–5. дои : 10.1038/nbt.1621 . ПМК 3146043 . ПМИД 20436464 .
- ^ Jump up to: а б Пахтер Л. (19 апреля 2011 г.). «Модели количественного определения транскриптов из RNA-Seq». arXiv : 1104.3889 [ q-bio.GN ].
- ^ «Что такое ФПКМ? Обзор единиц экспрессии RNA-Seq» . Фарраго . 8 мая 2014 года . Проверено 28 марта 2018 г.
- ^ Вагнер Г.П., Кин К., Линч В.Дж. (декабрь 2012 г.). «Измерение содержания мРНК с использованием данных секвенирования РНК: измерение RPKM непоследовательно в разных образцах». Теория в биологических науках . 131 (4): 281–5. дои : 10.1007/s12064-012-0162-3 . ПМИД 22872506 . S2CID 16752581 .
- ^ Эванс С., Хардин Дж., Стобель Д.М. (сентябрь 2018 г.). «Выбор методов нормализации межобразцовой РНК-Seq с точки зрения их предположений» . Брифинги по биоинформатике . 19 (5): 776–792. дои : 10.1093/нагрудник/bbx008 . ПМК 6171491 . ПМИД 28334202 .
- ^ Jump up to: а б Лоу К.В., Чен Ю, Ши В, Смит Г.К. (февраль 2014 г.). «voom: Прецизионные веса открывают инструменты анализа линейных моделей для подсчета считываний РНК-секвенирования» . Геномная биология . 15 (2): Р29. дои : 10.1186/gb-2014-15-2-r29 . ПМК 4053721 . ПМИД 24485249 .
- ^ Jump up to: а б Андерс С., Хубер В. (2010). «Дифференциальный экспрессионный анализ данных подсчета последовательностей» . Геномная биология . 11 (10): Р106. дои : 10.1186/gb-2010-11-10-r106 . ПМК 3218662 . ПМИД 20979621 .
- ^ Jump up to: а б Робинсон, доктор медицинских наук, Маккарти диджей, Смит Г.К. (январь 2010 г.). «edgeR: пакет Bioconductor для дифференциального анализа экспрессии цифровых данных об экспрессии генов» . Биоинформатика . 26 (1): 139–40. doi : 10.1093/биоинформатика/btp616 . ПМК 2796818 . ПМИД 19910308 .
- ^ Маргерат С., Шмидт А., Кодлин С., Чен В., Эберсольд Р., Бэлер Дж. (октябрь 2012 г.). «Количественный анализ транскриптомов и протеомов делящихся дрожжей в пролиферирующих и покоящихся клетках» . Клетка . 151 (3): 671–83. дои : 10.1016/j.cell.2012.09.019 . ПМЦ 3482660 . ПМИД 23101633 .
- ^ Оуэнс Н.Д., Блиц И.Л., Лейн М.А., Патрушев И., Овертон Дж.Д., Гилкрист М.Дж. и др. (январь 2016 г.). «Измерение абсолютного числа копий РНК с высоким временным разрешением показывает кинетику транскриптома в развитии» . Отчеты по ячейкам . 14 (3): 632–647. дои : 10.1016/j.celrep.2015.12.050 . ПМЦ 4731879 . ПМИД 26774488 .
- ^ Чен К., Ху Z, Ся Z, Чжао Д., Ли В., Тайлер Дж. К. (декабрь 2015 г.). «Упущенный факт: фундаментальная необходимость контроля всплесков практически для всех полногеномных анализов» . Молекулярная и клеточная биология . 36 (5): 662–7. дои : 10.1128/MCB.00970-14 . ПМК 4760223 . ПМИД 26711261 .
- ^ Ловен Дж., Орландо Д.А., Сигова А.А., Лин С.И., Рал П.Б., Бердж С.Б. и др. (октябрь 2012 г.). «Возвращаясь к глобальному анализу экспрессии генов» . Клетка . 151 (3): 476–82. дои : 10.1016/j.cell.2012.10.012 . ПМЦ 3505597 . ПМИД 23101621 .
- ^ Ричи М.Э., Фипсон Б., Ву Д., Ху Ю., Лоу К.В., Ши В. и др. (апрель 2015 г.). «Лимма обеспечивает дифференциальный анализ экспрессии для секвенирования РНК и исследований на микрочипах» . Исследования нуклеиновых кислот . 43 (7): е47. дои : 10.1093/нар/gkv007 . ПМК 4402510 . ПМИД 25605792 .
- ^ «Биокондуктор — программное обеспечение с открытым исходным кодом для биоинформатики» .
- ^ Хубер В., Кэри В.Дж., Джентльмен Р., Андерс С., Карлсон М., Карвальо Б.С. и др. (февраль 2015 г.). «Организация высокопроизводительного геномного анализа с помощью Bioconductor» . Природные методы . 12 (2): 115–21. дои : 10.1038/nmeth.3252 . ПМЦ 4509590 . ПМИД 25633503 .
- ^ Лик Дж.Т., Стори Дж.Д. (сентябрь 2007 г.). «Учет гетерогенности в исследованиях экспрессии генов с помощью анализа суррогатных переменных» . ПЛОС Генетика . 3 (9): 1724–35. дои : 10.1371/journal.pgen.0030161 . ЧВК 1994707 . ПМИД 17907809 .
- ^ Пиментел Х., Брэй Н.Л., Пуэнте С., Мелстед П., Пахтер Л. (июль 2017 г.). «Дифференциальный анализ РНК-секвенирования с учетом неопределенности количественного определения» . Природные методы . 14 (7): 687–690. дои : 10.1038/nmeth.4324 . ПМИД 28581496 . S2CID 15063247 .
- ^ Трапнелл С., Хендриксон Д.Г., Соважо М., Гофф Л., Ринн Дж.Л., Пахтер Л. (январь 2013 г.). «Дифференциальный анализ регуляции генов при разрешении транскриптов с помощью RNA-seq» . Природная биотехнология . 31 (1): 46–53. дои : 10.1038/nbt.2450 . ПМЦ 3869392 . ПМИД 23222703 .
- ^ Фрейзи А.С., Пертеа Дж., Джаффе А.Е., Лэнгмид Б., Зальцберг С.Л., Лик Дж.Т. (март 2015 г.). «Ballgown устраняет разрыв между сборкой транскриптома и анализом экспрессии» . Природная биотехнология . 33 (3): 243–6. дои : 10.1038/nbt.3172 . ПМЦ 4792117 . ПМИД 25748911 .
- ^ Jump up to: а б Сахрейян С.М., Мохиюддин М., Себра Р., Тилгнер Х., Афшар П.Т., Ау К.Ф. и др. (июль 2017 г.). «Получение всестороннего биологического понимания транскриптома путем выполнения анализа секвенирования РНК широкого спектра» . Природные коммуникации . 8 (1): 59. Бибкод : 2017NatCo...8...59S . дои : 10.1038/s41467-017-00050-4 . ПМЦ 5498581 . ПМИД 28680106 .
- ^ Циманн М., Эрен Ю., Эль-Оста А. (август 2016 г.). «Ошибки в названиях генов широко распространены в научной литературе» . Геномная биология . 17 (1): 177. дои : 10.1186/s13059-016-1044-7 . ПМЦ 4994289 . ПМИД 27552985 .
- ^ Сонесон С., Делоренци М. (март 2013 г.). «Сравнение методов дифференциального анализа экспрессии данных секвенирования РНК» . БМК Биоинформатика . 14:91 . дои : 10.1186/1471-2105-14-91 . ПМК 3608160 . ПМИД 23497356 .
- ^ Фонсека Н.А., Мариони Х., Бразма А. (30 сентября 2014 г.). «Профилирование генов RNA-Seq - систематическое эмпирическое сравнение» . ПЛОС ОДИН . 9 (9): e107026. Бибкод : 2014PLoSO...9j7026F . дои : 10.1371/journal.pone.0107026 . ПМЦ 4182317 . ПМИД 25268973 .
- ^ Сейеднасролла Ф., Лайхо А., Эло Л.Л. (январь 2015 г.). «Сравнение пакетов программного обеспечения для обнаружения дифференциальной экспрессии в исследованиях РНК-секвенирования» . Брифинги по биоинформатике . 16 (1): 59–70. дои : 10.1093/нагрудник/bbt086 . ПМЦ 4293378 . ПМИД 24300110 .
- ^ Рапапорт Ф., Ханин Р., Лян Й., Пирун М., Крек А., Зумбо П. и др. (2013). «Комплексная оценка методов анализа дифференциальной экспрессии генов для данных секвенирования РНК» . Геномная биология . 14 (9): 95 рандов. дои : 10.1186/gb-2013-14-9-r95 . ПМЦ 4054597 . ПМИД 24020486 .
- ^ Коста-Сильва Х., Домингес Д., Лопес FM (21 декабря 2017 г.). «Анализ дифференциальной экспрессии RNA-Seq: расширенный обзор и программный инструмент» . ПЛОС ОДИН . 12 (12): e0190152. Бибкод : 2017PLoSO..1290152C . дои : 10.1371/journal.pone.0190152 . ПМЦ 5739479 . ПМИД 29267363 .
- ^ Корчете Л.А., Рохас Э.А., Алонсо-Лопес Д., Де Лас Ривас Х., Гутьеррес Н.К., Бургильо Ф.Д. (12 ноября 2020 г.). «Систематическое сравнение и оценка процедур RNA-seq для количественного анализа экспрессии генов» . Научные отчеты . 12 (10): 19737. Бибкод : 2020NatSR..1019737C . дои : 10.1038/s41598-020-76881-x . ПМЦ 7665074 . ПМИД 33184454 .
- ^ Ляо И., Ван Дж., Джениг Э.Дж., Ши З., Чжан Б. (июль 2019 г.). «WebGestalt 2019: набор инструментов для анализа набора генов с обновленными пользовательскими интерфейсами и API» . Исследования нуклеиновых кислот . 47 (П1): И199–205. дои : 10.1093/nar/gkz401 . ПМК 6602449 . ПМИД 31114916 .
- ^ Jump up to: а б Керен Х., Лев-Маор Г., Аст Г. (май 2010 г.). «Альтернативный сплайсинг и эволюция: диверсификация, определение экзонов и функции». Обзоры природы. Генетика . 11 (5): 345–55. дои : 10.1038/nrg2776 . ПМИД 20376054 . S2CID 5184582 .
- ^ Лю Р., Лорейн А.Э., Дикерсон Дж.А. (декабрь 2014 г.). «Сравнение вычислительных методов дифференциального обнаружения альтернативного сплайсинга с использованием RNA-seq в растительных системах» . БМК Биоинформатика . 15 (1): 364. дои : 10.1186/s12859-014-0364-4 . ПМК 4271460 . ПМИД 25511303 .
- ^ Jump up to: а б Ли Й.И., Ноулз Д.А., Хамфри Дж., Барбейра А.Н., Дикинсон С.П., Им Х.К. и др. (январь 2018 г.). «Количественная оценка сплайсинга РНК без аннотаций с использованием LeafCutter» . Природная генетика . 50 (1): 151–158. дои : 10.1038/s41588-017-0004-9 . ПМК 5742080 . ПМИД 29229983 .
- ^ Андерс С., Рейес А., Хубер В. (октябрь 2012 г.). «Обнаружение дифференциального использования экзонов на основе данных РНК-секвенирования» . Геномные исследования . 22 (10): 2008–17. дои : 10.1101/гр.133744.111 . ПМК 3460195 . ПМИД 22722343 .
- ^ Шен С., Парк Дж.В., Хуан Дж., Диттмар К.А., Лу ZX, Чжоу Q и др. (апрель 2012 г.). «MATS: байесовский подход для гибкого обнаружения дифференциального альтернативного сплайсинга на основе данных RNA-Seq» . Исследования нуклеиновых кислот . 40 (8): е61. дои : 10.1093/nar/gkr1291 . ПМЦ 3333886 . ПМИД 22266656 .
- ^ Ван X, MJ Кэрнс (июнь 2014 г.). «SeqGSEA: пакет Bioconductor для анализа обогащения набора генов данными RNA-Seq, объединяющий дифференциальную экспрессию и сплайсинг» . Биоинформатика . 30 (12): 1777–9. doi : 10.1093/биоинформатика/btu090 . ПМИД 24535097 .
- ^ Трапнелл С., Хендриксон Д.Г., Соважо М., Гофф Л., Ринн Дж.Л., Пахтер Л. (январь 2013 г.). «Дифференциальный анализ регуляции генов при разрешении транскриптов с помощью RNA-seq» . Природная биотехнология . 31 (1): 46–53. дои : 10.1038/nbt.2450 . ПМЦ 3869392 . ПМИД 23222703 .
- ^ Ху Ю, Хуан Ю, Ду Ю, Орельяна С.Ф., Сингх Д., Джонсон А.Р. и др. (январь 2013 г.). «DiffSplice: полногеномное обнаружение событий дифференциального сплайсинга с помощью RNA-seq» . Исследования нуклеиновых кислот . 41 (2): е39. дои : 10.1093/нар/gks1026 . ПМЦ 3553996 . ПМИД 23155066 .
- ^ Вакеро-Гарсия Дж., Баррера А., Газзара М.Р., Гонсалес-Валлинас Дж., Лаэнс Н.Ф., Хогенеш Дж.Б. и др. (февраль 2016 г.). «Новый взгляд на сложность и регуляцию транскриптома через призму локальных вариаций сплайсинга» . электронная жизнь . 5 : е11752. дои : 10.7554/eLife.11752 . ПМК 4801060 . ПМИД 26829591 .
- ^ Мерино Г.А., Конеса А., Фернандес Э.А. (март 2019 г.). «Сравнительный анализ рабочих процессов для обнаружения дифференциального сплайсинга и дифференциальной экспрессии на уровне изоформ в исследованиях секвенирования РНК человека». Брифинги по биоинформатике . 20 (2): 471–481. дои : 10.1093/нагрудник/bbx122 . hdl : 11336/41247 . ПМИД 29040385 . S2CID 22706028 .
- ^ Jump up to: а б Маркотт Э.М., Пеллегрини М., Томпсон М.Дж., Йейтс Т.О., Айзенберг Д. (ноябрь 1999 г.). «Комбинированный алгоритм полногеномного прогнозирования функции белка». Природа . 402 (6757): 83–6. Бибкод : 1999Natur.402...83M . дои : 10.1038/47048 . ПМИД 10573421 . S2CID 144447 .
- ^ Jump up to: а б Георгий Ф.М., Дель Фаббро С., Ликаузи Ф. (март 2013 г.). «Сравнительное исследование сетей коэкспрессии, полученных на основе РНК-секвенирования и микрочипов, у Arabidopsis thaliana» . Биоинформатика . 29 (6): 717–24. doi : 10.1093/биоинформатика/btt053 . HDL : 11390/990155 . ПМИД 23376351 .
- ^ Янку О.Д., Каване С., Боттомли Д., Сирлз Р., Хитземанн Р., МакВини С. (июнь 2012 г.). «Использование данных RNA-Seq для вывода о сети коэкспрессии de novo» . Биоинформатика . 28 (12): 1592–7. doi : 10.1093/биоинформатика/bts245 . ПМЦ 3493127 . ПМИД 22556371 .
- ^ Экси Р., Ли Х.Д., Менон Р., Вэнь Ю., Оменн Г.С., Крецлер М. и др. (ноябрь 2013 г.). «Систематическое дифференцирование функций альтернативно сплайсированных изоформ посредством интеграции данных секвенирования РНК» . PLOS Вычислительная биология . 9 (11): e1003314. Бибкод : 2013PLSCB...9E3314E . дои : 10.1371/journal.pcbi.1003314 . ПМЦ 3820534 . ПМИД 24244129 .
- ^ Ли Х.Д., Менон Р., Оменн Г.С., Гуань Ю (август 2014 г.). «Новая эра интеграции геномных данных для анализа функции изоформ сплайсинга» . Тенденции в генетике . 30 (8): 340–7. дои : 10.1016/j.tig.2014.05.005 . ПМЦ 4112133 . ПМИД 24951248 .
- ^ Форушани А., Аграхари Р., Докинг Р., Чанг Л., Дунс Г., Худоба М. и др. (март 2017 г.). «Крупномасштабный анализ генной сети показывает значение пути внеклеточного матрикса и гомеобоксных генов при остром миелоидном лейкозе: введение в пакет Pigengene и его применение» . BMC Медицинская Геномика . 10 (1): 16. дои : 10.1186/s12920-017-0253-6 . ПМЦ 5353782 . ПМИД 28298217 .
- ^ Ли Х., Рукосакер Б., Вайсокер А., Феннелл Т., Руан Дж., Гомер Н. и др. (август 2009 г.). «Формат Sequence Alignment/Map и SAMtools» . Биоинформатика . 25 (16): 2078–9. doi : 10.1093/биоинформатика/btp352 . ПМК 2723002 . ПМИД 19505943 .
- ^ ДеПристо М.А., Бэнкс Э., Поплин Р., Гаримелла К.В., Магуайр Дж.Р., Хартл С. и др. (май 2011 г.). «Система обнаружения вариаций и генотипирования с использованием данных секвенирования ДНК нового поколения» . Природная генетика . 43 (5): 491–8. дои : 10.1038/ng.806 . ПМК 3083463 . ПМИД 21478889 .
- ^ Battle A, Brown CD, Энгельхардт BE, Монтгомери SB (октябрь 2017 г.). «Генетическое влияние на экспрессию генов в тканях человека» . Природа . 550 (7675): 204–213. Бибкод : 2017Natur.550..204A . дои : 10.1038/nature24277 . hdl : 10230/34202 . ПМК 5776756 . ПМИД 29022597 .
- ^ Рихтер Ф., Хоффман Г.Э., Манхаймер К.Б., Патель Н., Шарп А.Дж., МакКин Д. и др. (октябрь 2019 г.). «ORE определяет крайние эффекты экспрессии, обогащенные редкими вариантами» . Биоинформатика . 35 (20): 3906–3912. doi : 10.1093/биоинформатика/btz202 . ПМК 6792115 . ПМИД 30903145 .
- ^ Фридман А.Х., Клэмп М., Сактон ТБ (январь 2021 г.). «Ошибка, шум и предвзятость в сборках транскриптома de novo». Ресурсы молекулярной экологии . 21 (1): 18–29. дои : 10.1111/1755-0998.13156 . ПМИД 32180366 . S2CID 212739959 .
- ^ Тейшейра М.Р. (декабрь 2006 г.). «Рецидивирующие онкогены слияния при карциномах». Критические обзоры онкогенеза . 12 (3–4): 257–71. дои : 10.1615/critrevoncog.v12.i3-4.40 . ПМИД 17425505 . S2CID 40770452 .
- ^ Тинд А.С., Монга И., Тхакур П.К., Кумари П., Диндория К., Крзак М. и др. (ноябрь 2021 г.). «Демистификация новых приложений массового секвенирования РНК: применение и полезность биоинформатической методологии». Брифинги по биоинформатике . 22 (6). дои : 10.1093/нагрудник/bbab259 . ПМИД 34329375 .
- ^ Санджованни М., Граната И., Тинд А.С., Гуаррачино М.Р. (апрель 2019 г.). «От мусора к сокровищу: обнаружение неожиданного загрязнения в некартированных данных NGS» . БМК Биоинформатика . 20 (Приложение 4): 168. doi : 10.1186/s12859-019-2684-x . ПМК 6472186 . ПМИД 30999839 .
- ^ «Поиск в PubMed: «RNA Seq» ИЛИ «RNA-Seq» ИЛИ «Секвенирование РНК» ИЛИ «RNASeq» » . ПабМед . Проверено 20 июня 2021 г.
- ^ «Поиск в PubMed: («RNA Seq» ИЛИ «RNA-Seq» ИЛИ «Секвенирование РНК» ИЛИ «RNASeq») И «Медицина» " . ПабМед . Проверено 20 июня 2021 г.
- ^ Вебер А.П. (ноябрь 2015 г.). «Открытие новой биологии посредством секвенирования РНК» . Физиология растений . 169 (3): 1524–31. дои : 10.1104/стр.15.01081 . ПМК 4634082 . ПМИД 26353759 .
- ^ Бейнбридж М.Н., Уоррен Р.Л., Херст М., Романуик Т., Зенг Т., Го А. и др. (сентябрь 2006 г.). «Анализ транскриптома линии клеток рака простаты LNCaP с использованием подхода секвенирования путем синтеза» . БМК Геномика . 7 : 246. дои : 10.1186/1471-2164-7-246 . ПМЦ 1592491 . ПМИД 17010196 .
- ^ Чунг Ф., Хаас Б.Дж., Голдберг С.М., Мэй Г.Д., Сяо Ю., Town CD (октябрь 2006 г.). «Секвенирование Medicago truncatula экспрессировало секвенированные теги с использованием технологии 454 Life Sciences» . БМК Геномика . 7 : 272. дои : 10.1186/1471-2164-7-272 . ПМК 1635983 . ПМИД 17062153 .
- ^ Эмрих С.Дж., Барбазук В.Б., Ли Л., Шнабле П.С. (январь 2007 г.). «Открытие и аннотация генов с использованием секвенирования транскриптома LCM-454» . Геномные исследования . 17 (1): 69–73. дои : 10.1101/гр.5145806 . ПМК 1716268 . ПМИД 17095711 .
- ^ Вебер А.П., Вебер К.Л., Карр К., Вилкерсон С., Ольрогге Дж.Б. (май 2007 г.). «Отбор проб транскриптома Arabidopsis с помощью массового параллельного пиросеквенирования» . Физиология растений . 144 (1): 32–42. дои : 10.1104/стр.107.096677 . ЧВК 1913805 . ПМИД 17351049 .
- ^ Нагалакшми У., Ван З., Ваерн К., Шу С., Раха Д., Герштейн М. и др. (июнь 2008 г.). «Транскрипционный ландшафт генома дрожжей, определенный с помощью секвенирования РНК» . Наука . 320 (5881): 1344–9. Бибкод : 2008Sci...320.1344N . дои : 10.1126/science.1158441 . ПМЦ 2951732 . ПМИД 18451266 .
- ^ Рихтер Ф (2021). «Широкое введение в RNA-Seq» . Викижурнал науки . 4 (1): 4. doi : 10.15347/WJS/2021.004 .
- ^ Каммингс Б.Б., Маршалл Дж.Л., Тукиайнен Т., Лек М., Донкерворт С., Фоли А.Р. и др. (19 апреля 2017 г.). «Улучшение генетической диагностики менделевской болезни с помощью секвенирования транскриптома». Наука трансляционной медицины . 9 (386). doi : 10.1126/scitranslmed.aal5209 . hdl : 10230/35912 .
- ^ Кремер Л.С., Бадер Д.М., Мертес С., Копайтич Р., Пихлер Г., Юсо А. и др. (12 июня 2017 г.). «Генетическая диагностика менделевских расстройств с помощью секвенирования РНК». Природные коммуникации . 8 (1). дои : 10.1038/ncomms15824 .
- ^ Сандберг Р. (январь 2014 г.). «Вход в эпоху одноклеточной транскриптомики в биологии и медицине» . Природные методы . 11 (1): 22–4. дои : 10.1038/nmeth.2764 . ПМИД 24524133 . S2CID 27632439 .
- ^ «КОДИРОВАТЬ матрицу данных» . Проверено 28 июля 2013 г.
- ^ «Атлас генома рака - Портал данных» . Проверено 28 июля 2013 г.
Дальнейшее чтение
[ редактировать ]- Тагучи Ю. (2019). «Сравнительный транскриптомический анализ». Энциклопедия биоинформатики и вычислительной биологии . стр. 814–818. дои : 10.1016/B978-0-12-809633-8.20163-5 . ISBN 978-0-12-811432-2 . S2CID 65302519 .
Внешние ссылки
[ редактировать ]- Креско Б., Фёлкер Р., Смолл С. (2001). Башем С., Кэтчен Дж. (ред.). «РНК-секлопедия» . Университет Орегона. : общее руководство по планированию и проведению эксперимента по секвенированию РНК.