Фон Хиппель -Линдау болезнь
| Фон Хиппель -Линдау болезнь | |
|---|---|
| Другие имена | Семейный ангиоматоз сетчатки мозга [ 1 ] |
 | |
| Места основных типов кист и опухолей при болезни фон Хиппель -Линдау. [ 2 ] | |
| Специальность | Медицинская генетика , неврология |
Болезнь фон Хиппель -Линдау ( VHL ), также известная как синдром фон Хиппель -Линдау , является редким генетическим заболеванием с участием мультисистемы. [ 3 ] Он характеризуется висцеральными кистами и доброкачественными опухолями с потенциалом для последующей злокачественной трансформации. Это тип факоматоза , который является результатом мутации в ген -супрессоре опухоли фон Хиппель -Линдау на хромосоме 3p 25.3. [ 4 ] [ 5 ] [ 6 ]
Признаки и симптомы
[ редактировать ]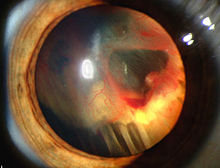
| Опухоль/тип кисты [ 2 ] | Распространенность [ 2 ] |
|---|---|
| Поджелудочная киста | 50-91% |
| Мозжечок гемангиобластома | 44-72% |
| Почечные кисты | 59-63% |
| Гемангиобластома сетчатки | 45-59% |
| Почечный рак | 24-45% |
| Спинной мозг гемангиобластома | 13-59% |
| Папиллярная цистаденома эпидидимиса | 10-60% мужчин |
| Феохромоцитома | 0-60% |
| Нейроэндокринная опухоль поджелудочной железы | 5-17% |
| Серозная цистаденома поджелудочной железы | 12% |
| Медуллярная гемангиобластома | 5% |

Признаки и симптомы, связанные с болезнью ВХЛ, включают в себя головные боли, проблемы с балансом и ходьбой, головокружение, слабость конечностей, проблемы зрения и высокое кровяное давление.
Условия, связанные с заболеванием ВХЛ, включают ангиоматоз , гемангиобластомы , феохромоцитому , почечной карциномы , поджелудочной кисты железы ( поджелудочная серозная цистаденома ), эндолимфатический мешок и двусторонний папиллярный цистаденомы эпидидима мужчин ) ( . [ 7 ] [ 8 ] Ангиоматоз возникает у 37,2% пациентов с заболеванием ВХЛ и обычно встречается в сетчатке. В результате потеря зрения очень распространена. Однако могут быть затронуты другие органы: инсульты, сердечные приступы и сердечно -сосудистые заболевания являются распространенными дополнительными симптомами. [6] Approximately 40% of VHL disease presents with CNS hemangioblastomas and they are present in around 60–80%. Spinal hemangioblastomas are found in 13–59% of VHL disease and are specific because 80% are found in VHL disease.[9][10] Although all of these tumors are common in VHL disease, around half of cases present with only one tumor type.[10] Most people with VHL develop symptoms in their mid-twenties.[11]
Pathogenesis
[edit]The disease is caused by mutations of the Von Hippel–Lindau tumor suppressor (VHL) gene on the short arm of chromosome 3 (3p25-26). There are over 1500 germline mutations and somatic mutations found in VHL disease.[12][13]

Every cell in the body has two copies of every gene (bar those found in the sex chromosomes, X and Y). In VHL disease, one copy of the VHL gene has a mutation and produces a faulty VHL protein (pVHL). However, the second copy still produces a functional protein. The condition is inherited in an autosomal dominant manner – one copy of the faulty gene is sufficient to increase the risk of developing tumours.[14][15]
Approximately 20% of cases of VHL disease are found in individuals without a family history, known as de novo mutations. An inherited mutation of the VHL gene is responsible for the remaining 80 percent of cases.[9]
30–40% of mutations in the VHL gene consist of 50-250kb deletion mutations that remove either part of the gene or the whole gene and flanking regions of DNA. The remaining 60-70% of VHL disease is caused by the truncation of pVHL by nonsense mutations, indel mutations or splice site mutations.[9]
VHL protein
[edit]
The VHL protein (pVHL) is involved in the regulation of a protein known as hypoxia inducible factor 1α (HIF1α). This is a subunit of a heterodimeric transcription factor that at normal cellular oxygen levels is highly regulated. In normal physiological conditions, pVHL recognizes and binds to HIF1α only when oxygen is present due to the post translational hydroxylation of 2 proline residues within the HIF1α protein. pVHL is an E3 ligase that ubiquitinates HIF1α and causes its degradation by the proteasome. In low oxygen conditions or in cases of VHL disease where the VHL gene is mutated, pVHL does not bind to HIF1α. This allows the subunit to dimerise with HIF1β and activate the transcription of a number of genes, including vascular endothelial growth factor, platelet-derived growth factor B, erythropoietin and genes involved in glucose uptake and metabolism.[15][16] A new novel missense mutation in VHL genes c.194 C>T, c.239 G>A, c.278 G>A, c.319 C>G, c.337 C>G leading to the following variations p.Ala 65 Val, p.Gly 80 Asp, p.Gly 93 Glu, p.Gln 107 Glu, p.Gln 113 Glu [check spelling] in the protein contributed to renal clear cell carcinoma.[17]
Diagnosis
[edit]The detection of tumours specific to VHL disease is important in the disease's diagnosis. In individuals with a family history of VHL disease, one hemangioblastoma, pheochromocytoma or renal cell carcinoma may be sufficient to make a diagnosis. As all the tumours associated with VHL disease can be found sporadically, at least two tumours must be identified to diagnose VHL disease in a person without a family history.[9][10]
Genetic diagnosis is also useful in VHL disease diagnosis. In hereditary VHL disease, techniques such as the Southern blot and gene sequencing can be used to analyse DNA and identify mutations. These tests can be used to screen family members of those afflicted with VHL disease; de novo cases that produce genetic mosaicism are more difficult to detect because mutations are not found in the white blood cells that are used for genetic analysis.[9][18]
Classification
[edit]VHL disease can be subdivided according to the clinical manifestations, although these groups often correlate with certain types of mutations present in the VHL gene.[19]
Treatment
[edit]Early recognition and treatment of specific manifestations of VHL can substantially decrease complications and improve quality of life. For this reason, individuals with VHL disease are usually screened routinely for retinal angiomas, CNS hemangioblastomas, clear-cell renal carcinomas and pheochromocytomas.[20] CNS hemangioblastomas are usually surgically removed if they are symptomatic. Photocoagulation and cryotherapy are usually used for the treatment of symptomatic retinal angiomas, although anti-angiogenic treatments may also be an option. Renal tumours may be removed by a partial nephrectomy or other techniques such as radiofrequency ablation.[9]
Belzutifan is a drug under investigation for the treatment of von Hippel–Lindau disease-associated renal cell carcinoma.[21][22][23][24]
Epidemiology
[edit]VHL disease has an incidence of one in 36,000 births. There is over 90% penetrance by the age of 65.[25] Age at diagnosis varies from infancy to age 60–70 years, with an average patient age at clinical diagnosis of 26 years.[citation needed]
History
[edit]
The German ophthalmologist Eugen von Hippel first described angiomas in the eye in 1904.[26] Arvid Lindau described the angiomas of the cerebellum and spine in 1927.[27] The term Von Hippel–Lindau disease was first used in 1936; however, its use became common only in the 1970s.[9]
Notable cases
[edit]Some descendants of the McCoy family (involved in the Hatfield-McCoy feud of Appalachia, USA) are presumed to have VHL. In an article appearing in the Associated Press, it has been speculated by a Vanderbilt University endocrinologist that the hostility underlying the Hatfield–McCoy feud may have been partly due to the consequences of Von Hippel–Lindau disease. The article suggests that the McCoy family was predisposed to bad tempers because many of them had a pheochromocytoma that produced excess adrenaline and a tendency toward explosive tempers.[28] An update of the Associated Press article in 2023 carries more details.[29]
Nomenclature
[edit]Other uncommon names are: angiomatosis retinae, familial cerebello-retinal angiomatosis, cerebelloretinal hemangioblastomatosis, Hippel Disease, Hippel–Lindau syndrome, HLS, VHL, Lindau disease or retinocerebellar angiomatosis.[30][31]
See also
[edit]References
[edit]- ^ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Von Hippel Lindau disease". www.orpha.net. Retrieved 25 May 2019.
{{cite web}}: CS1 maint: numeric names: authors list (link) - ^ Jump up to: a b c Leung RS, Biswas SV, Duncan M, Rankin S (2008). "Imaging features of von Hippel-Lindau disease". Radiographics. 28 (1): 65–79, quiz 323. doi:10.1148/rg.281075052. PMID 18203931.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ "Von Hippel-Lindau disease | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2018-04-17.
- ^ Richard, S; Gardie, B; Couvé, S; Gad, S (May 30, 2012). "Von Hippel-Lindau: How a rare disease illuminates cancer biology". Seminars in Cancer Biology. 23 (1): 26–37. doi:10.1016/j.semcancer.2012.05.005. PMID 22659535. Archived from the original (PDF) on April 9, 2022. Retrieved April 20, 2018.
- ^ Henry, Todd; Campell, James; Hawley, Arthur (1969). Todd-Sanford clinical diagnosis by laboratory methods, edited by Israel Davidsohn [and] John Bernard Henry (14th ed.). Philadelphia: Saunders. p. 555. ISBN 978-0-7216-2921-6.
- ^ Jump up to: a b Wong WT; Agrón E; Coleman HR; et al. (February 2007). "Genotype–phenotype correlation in von Hippel–Lindau disease with retinal angiomatosis". Archives of Ophthalmology. 125 (2): 239–45. doi:10.1001/archopht.125.2.239. PMC 3019103. PMID 17296901. Archived from the original on 2008-12-12. Retrieved 2008-10-22.
- ^ Lindsay, Kenneth W; Ian Bone; Robin Callander; J. van Gijn (1991). Neurology and Neurosurgery Illustrated. United States: Churchill Livingstone. ISBN 978-0-443-04345-1.
- ^ Frantzen, Carlijn; Links, Thera P.; Giles, Rachel H. (21 June 2012). "Von Hippel-Lindau Syndrome". Von Hippel-Lindau Disease. University of Washington, Seattle. PMID 20301636. Retrieved 30 March 2013.
{{cite book}}:|work=ignored (help) - ^ Jump up to: a b c d e f g Maher ER; Glenn GM; Walther M; et al. (June 2011). "von Hippel-Lindau disease: a clinical and scientific review". European Journal of Human Genetics. 19 (6): 617–23. doi:10.1038/ejhg.2010.175. PMC 3110036. PMID 21386872.
- ^ Jump up to: a b c Friedrich, CA (December 1, 1999). "Von Hippel-Lindau syndrome. A pleomorphic condition". Cancer. 86 (11 Suppl): 2478–82. doi:10.1002/(SICI)1097-0142(19991201)86:11+<2478::AID-CNCR4>3.0.CO;2-5. PMID 10630173. S2CID 45672391.
- ^ Clinic, Cleveland (2017-10-16). "Von Hippel-Lindau Disease (VHL)". Cleveland Clinic.
- ^ Kondo, K; Kaelin Jr, WG (10 March 2001). "The von Hippel–Lindau Tumor Suppressor Gene". Experimental Cell Research. 264 (1): 117–125. doi:10.1006/excr.2000.5139. PMID 11237528.
- ^ Nordstrom-O'Brien M; van der Luijt RB; van Rooijen E; et al. (May 2010). "Genetic analysis of von Hippel-Lindau disease". Hum. Mutat. 31 (5): 521–37. doi:10.1002/humu.21219. PMID 20151405. S2CID 38910112.
- ^ Knudson, AG (Nov 2001). "Two genetic hits (more or less) to cancer". Nature Reviews Cancer. 1 (2): 157–62. doi:10.1038/35101031. PMID 11905807. S2CID 20201610.
- ^ Jump up to: a b Kaelin, WG (2007). "Von Hippel-Lindau disease". Annual Review of Pathology. 2: 145–73. doi:10.1146/annurev.pathol.2.010506.092049. PMID 18039096.
- ^ Bader, HL; Hsu, T (June 4, 2012). "Systemic VHL gene functions and the VHL disease". FEBS Letters. 586 (11): 1562–9. doi:10.1016/j.febslet.2012.04.032. PMC 3372859. PMID 22673568.
- ^ Kumar, P. S.; Venkatesh, K.; Srikanth, L.; Sarma, P. V.; Reddy, A. R.; Subramanian, S.; Phaneendra, B. V. (July 2013). "Novel three missense mutations observed in Von Hippel-Lindau gene in a patient reported with renal cell carcinoma". Indian Journal of Human Genetics. 19 (3): 373–376. doi:10.4103/0971-6866.120809. PMC 3841571. PMID 24339559.
- ^ Lonser RR (June 2003). "von Hippel-Lindau disease". Lancet. 361 (9374): 2059–67. doi:10.1016/S0140-6736(03)13643-4. PMID 12814730. S2CID 13783714.
- ^ Calzada, MJ (March 2010). "Von Hippel-Lindau syndrome: molecular mechanisms of the disease". Clinical & Translational Oncology. 12 (3): 160–5. doi:10.1007/s12094-010-0485-9. PMID 20231120. S2CID 7789108.
- ^ Priesemann M; Davies KM; Perry LA; et al. (2006). "Benefits of screening in von Hippel-Lindau disease – comparison of morbidity associated with initial tumours in affected parents and children". Horm. Res. 66 (1): 1–5. doi:10.1159/000093008. PMID 16651847. S2CID 29862078.
- ^ "Belzutifan". SPS - Specialist Pharmacy Service. 18 March 2021. Archived from the original on 26 April 2021. Retrieved 25 April 2021.
- ^ «MHRA Awards First« Passport »под новым путем» . Рэп (пресс -релиз) . Получено 25 апреля 2021 года .
- ^ «Merck получает приоритетный обзор от FDA для нового применения лекарств для ингибитора HIF-2α Belzutifan (MK-6482)» (пресс-релиз). Мерк. 16 марта 2016 года . Получено 25 апреля 2021 года - через Business Wire.
- ^ «FDA предоставляет приоритетный обзор Belzutifan для RCC, связанного с болезнью Фон Хиппель-Линдау» . Раковая сеть . 16 марта 2021 года . Получено 2021-04-26 .
- ^ Ким, JJ; Рини, Би; Hansel, DE (2010). «Синдром фон Хиппель Линдау». Заболевания репарации ДНК . Достижения в области экспериментальной медицины и биологии. Тол. 685. С. 228–49. doi : 10.1007/978-1-4419-6448-9_22 . ISBN 978-1-4419-6447-2 Полем PMID 20687511 .
- ^ Хиппель Э (1904). «О очень редкой болезни сетчатки» . Архив Альбрехта фон Грэфе для офтальмологии . 59 : 83-106. Doi : 10.1007/bf01994821 . S2CID 22425158 .
- ^ Линдау А (1927). «На вопрос об ангиоматозе сетчатка и ее осложнения в мозге». Acta Ophthalmologica . 4 (1–2): 193–226. Doi : 10.1111/j.1755-3768.1926.tb07786.x . S2CID 73385451 .
- ^ « Хэтфилд -Мккойская Федя обвиняла в« ярости »болезни » . Msnbc.com. 2007-04-05. Архивировано из оригинала 2007-04-07 . Получено 2007-04-05 .
- ^ Маркионе, Мэрилинн; Breed, Allen G., вражда Hatfield-McCoy может быть частично объяснена редким заболеванием, которое вызывает ярость , австралийская пресса (AP), 4 августа 2023 г.
- ^ «Болезнь фон Хиппель-Линдау» . База данных редких заболеваний . Норд - Национальная организация для редких расстройств. 2019. Архивировано с оригинала 25 апреля 2015 года . Получено 4 апреля 2022 года .
- ^ «Болезнь фон Хиппель-Линдау» . Медицинские заголовки (сетка) . Национальная библиотека медицины США . Получено 4 апреля 2022 года .
Внешние ссылки
[ редактировать ]- Generons/NCBI/NIH/ваша запись на синдроме фон Хиппель-Линдау
- Фон Хиппель -Линдау болезнь (VHL) в Ninds
- Синдром фон Хиппель -Линдау в NLM Genetics Home Ссылка
- Болезнь Хиппеля -Линдау в том, кто его назвал?
- Онлайн -наследство Менделян в Человеке (OMIM): 608537 (ген VHL)