Альтернативный сплайсинг

Альтернативный сплайсинг , или альтернативный сплайсинг РНК , или дифференциальный сплайсинг , представляют собой альтернативный процесс сплайсинга во время экспрессии генов, который позволяет одному геном продуцировать различные варианты сплайсинга. Например, некоторые экзоны гена могут быть включены в или исключены из окончательного РНК -продукта гена. [ 1 ] Это означает, что экзоны соединяются в разных комбинациях, что приводит к различным вариантам сплайсинга. В случае генов, кодирующих белок, белки, переведенные из этих вариантов сплайсинга, могут содержать различия в их аминокислотной последовательности и в их биологических функциях (см. Рисунок).
Биологически значимый альтернативный сплайсинг происходит в качестве нормального явления у эукариот , где он увеличивает количество белков, которые могут кодироваться геномом. [ 1 ] У людей широко распространено мнение, что ~ 95% мультиэкс-генов альтернативно сплайсированы для производства функциональных альтернативных продуктов из одного и того же гена [ 2 ] but many scientists believe that most of the observed splice variants are due to splicing errors and the actual number of biologically relevant alternatively spliced genes is much lower.[3][4]
Discovery
[edit]Alternative splicing was first observed in 1977.[5][6] The adenovirus produces five primary transcripts early in its infectious cycle, prior to viral DNA replication, and an additional one later, after DNA replication begins. The early primary transcripts continue to be produced after DNA replication begins. The additional primary transcript produced late in infection is large and comes from 5/6 of the 32kb adenovirus genome. This is much larger than any of the individual adenovirus mRNAs present in infected cells. Researchers found that the primary RNA transcript produced by adenovirus type 2 in the late phase was spliced in many different ways, resulting in mRNAs encoding different viral proteins. In addition, the primary transcript contained multiple polyadenylation sites, giving different 3' ends for the processed mRNAs.[7][8][9]
In 1981, the first example of alternative splicing in a transcript from a normal, endogenous gene was characterized.[7] The gene encoding the thyroid hormone calcitonin was found to be alternatively spliced in mammalian cells. The primary transcript from this gene contains 6 exons; the calcitonin mRNA contains exons 1–4, and terminates after a polyadenylation site in exon 4. Another mRNA is produced from this pre-mRNA by skipping exon 4, and includes exons 1–3, 5, and 6. It encodes a protein known as CGRP (calcitonin gene related peptide).[10][11] Examples of alternative splicing in immunoglobin gene transcripts in mammals were also observed in the early 1980s.[7][12]
Since then, many other examples of biologically relevant alternative splicing have been found in eukaryotes.[1] The "record-holder" for alternative splicing is a D. melanogaster gene called Dscam, which could potentially have 38,016 splice variants.[13]
In 2021, it was discovered that the genome of adenovirus type 2, the adenovirus in which alternative splicing was first identified, was able to produce a much greater variety of splice variants than previously thought.[14] By using next generation sequencing technology, researchers were able to update the human adenovirus type 2 transcriptome and document the presence of 904 splice variants produced by the virus through a complex pattern of alternative splicing. Very few of these splice variants have been shown to be functional, a point that the authors raise in their paper.
- "An outstanding question is what roles the menagerie of novel RNAs play or whether they are spurious molecules generated by an overloaded splicing machinery."[14]
Modes
[edit]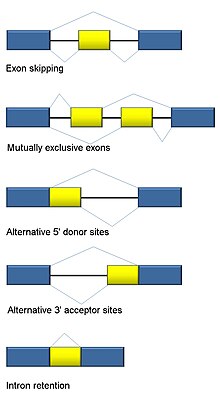

Five basic modes of alternative splicing are generally recognized.[1][2][16][15]
- Exon skipping or cassette exon: in this case, an exon may be spliced out of the primary transcript or retained. This is the most common mode in mammalian pre-mRNAs.[15]
- Mutually exclusive exons: One of two exons is retained in mRNAs after splicing, but not both.
- Alternative donor site: An alternative 5' splice junction (donor site) is used, changing the 3' boundary of the upstream exon.
- Alternative acceptor site: An alternative 3' splice junction (acceptor site) is used, changing the 5' boundary of the downstream exon.
- Intron retention: A sequence may be spliced out as an intron or simply retained. This is distinguished from exon skipping because the retained sequence is not flanked by introns. If the retained intron is in the coding region, the intron must encode amino acids in frame with the neighboring exons, or a stop codon or a shift in the reading frame will cause the protein to be non-functional. This is the rarest mode in mammals but the most common in plants.[15][17]
In addition to these primary modes of alternative splicing, there are two other main mechanisms by which different mRNAs may be generated from the same gene; multiple promoters and multiple polyadenylation sites. Use of multiple promoters is properly described as a transcriptional regulation mechanism rather than alternative splicing; by starting transcription at different points, transcripts with different 5'-most exons can be generated. At the other end, multiple polyadenylation sites provide different 3' end points for the transcript. Both of these mechanisms are found in combination with alternative splicing and provide additional variety in mRNAs derived from a gene.[1][16]

These modes describe basic splicing mechanisms, but may be inadequate to describe complex splicing events. For instance, the figure to the right shows 3 spliceforms from the mouse hyaluronidase 3 gene. Comparing the exonic structure shown in the first line (green) with the one in the second line (yellow) shows intron retention, whereas the comparison between the second and the third spliceform (yellow vs. blue) exhibits exon skipping. A model nomenclature to uniquely designate all possible splicing patterns has recently been proposed.[15]
Mechanisms
[edit]General splicing mechanism
[edit]
When the pre-mRNA has been transcribed from the DNA, it includes several introns and exons. (In nematodes, the mean is 4–5 exons and introns; in the fruit fly Drosophila there can be more than 100 introns and exons in one transcribed pre-mRNA.) The exons to be retained in the mRNA are determined during the splicing process. The regulation and selection of splice sites are done by trans-acting splicing activator and splicing repressor proteins as well as cis-acting elements within the pre-mRNA itself such as exonic splicing enhancers and exonic splicing silencers.
The typical eukaryotic nuclear intron has consensus sequences defining important regions. Each intron has the sequence GU at its 5' end. Near the 3' end there is a branch site. The nucleotide at the branchpoint is always an A; the consensus around this sequence varies somewhat. In humans the branch site consensus sequence is yUnAy.[18] The branch site is followed by a series of pyrimidines – the polypyrimidine tract – then by AG at the 3' end.[16]
Splicing of mRNA is performed by an RNA and protein complex known as the spliceosome, containing snRNPs designated U1, U2, U4, U5, and U6 (U3 is not involved in mRNA splicing).[19] U1 binds to the 5' GU and U2, with the assistance of the U2AF protein factors, binds to the branchpoint A within the branch site. The complex at this stage is known as the spliceosome A complex. Formation of the A complex is usually the key step in determining the ends of the intron to be spliced out, and defining the ends of the exon to be retained.[16] (The U nomenclature derives from their high uridine content).
The U4,U5,U6 complex binds, and U6 replaces the U1 position. U1 and U4 leave. The remaining complex then performs two transesterification reactions. In the first transesterification, 5' end of the intron is cleaved from the upstream exon and joined to the branch site A by a 2',5'-phosphodiester linkage. In the second transesterification, the 3' end of the intron is cleaved from the downstream exon, and the two exons are joined by a phosphodiester bond. The intron is then released in lariat form and degraded.[1]
Regulatory elements and proteins
[edit]
Splicing is regulated by trans-acting proteins (repressors and activators) and corresponding cis-acting regulatory sites (silencers and enhancers) on the pre-mRNA. However, as part of the complexity of alternative splicing, it is noted that the effects of a splicing factor are frequently position-dependent. That is, a splicing factor that serves as a splicing activator when bound to an intronic enhancer element may serve as a repressor when bound to its splicing element in the context of an exon, and vice versa.[20] The secondary structure of the pre-mRNA transcript also plays a role in regulating splicing, such as by bringing together splicing elements or by masking a sequence that would otherwise serve as a binding element for a splicing factor.[21][22] Together, these elements form a "splicing code" that governs how splicing will occur under different cellular conditions.[23][24]
There are two major types of cis-acting RNA sequence elements present in pre-mRNAs and they have corresponding trans-acting RNA-binding proteins. Splicing silencers are sites to which splicing repressor proteins bind, reducing the probability that a nearby site will be used as a splice junction. These can be located in the intron itself (intronic splicing silencers, ISS) or in a neighboring exon (exonic splicing silencers, ESS). They vary in sequence, as well as in the types of proteins that bind to them. The majority of splicing repressors are heterogeneous nuclear ribonucleoproteins (hnRNPs) such as hnRNPA1 and polypyrimidine tract binding protein (PTB).[16][23] Splicing enhancers are sites to which splicing activator proteins bind, increasing the probability that a nearby site will be used as a splice junction. These also may occur in the intron (intronic splicing enhancers, ISE) or exon (exonic splicing enhancers, ESE). Most of the activator proteins that bind to ISEs and ESEs are members of the SR protein family. Such proteins contain RNA recognition motifs and arginine and serine-rich (RS) domains.[16][23]

In general, the determinants of splicing work in an inter-dependent manner that depends on context, so that the rules governing how splicing is regulated form a splicing code.[24] The presence of a particular cis-acting RNA sequence element may increase the probability that a nearby site will be spliced in some cases, but decrease the probability in other cases, depending on context. The context within which regulatory elements act includes cis-acting context that is established by the presence of other RNA sequence features, and trans-acting context that is established by cellular conditions. For example, some cis-acting RNA sequence elements influence splicing only if multiple elements are present in the same region so as to establish context. As another example, a cis-acting element can have opposite effects on splicing, depending on which proteins are expressed in the cell (e.g., neuronal versus non-neuronal PTB). The adaptive significance of splicing silencers and enhancers is attested by studies showing that there is strong selection in human genes against mutations that produce new silencers or disrupt existing enhancers.[25][26]
Examples
[edit]Exon skipping: Drosophila dsx
[edit]
Pre-mRNAs from the D. melanogaster gene dsx contain 6 exons. In males, exons 1,2,3,5,and 6 are joined to form the mRNA, which encodes a transcriptional regulatory protein required for male development. In females, exons 1,2,3, and 4 are joined, and a polyadenylation signal in exon 4 causes cleavage of the mRNA at that point. The resulting mRNA is a transcriptional regulatory protein required for female development.[27]
This is an example of exon skipping. The intron upstream from exon 4 has a polypyrimidine tract that doesn't match the consensus sequence well, so that U2AF proteins bind poorly to it without assistance from splicing activators. This 3' splice acceptor site is therefore not used in males. Females, however, produce the splicing activator Transformer (Tra) (see below). The SR protein Tra2 is produced in both sexes and binds to an ESE in exon 4; if Tra is present, it binds to Tra2 and, along with another SR protein, forms a complex that assists U2AF proteins in binding to the weak polypyrimidine tract. U2 is recruited to the associated branchpoint, and this leads to inclusion of exon 4 in the mRNA.[27][28]
Alternative acceptor sites: Drosophila Transformer
[edit]Pre-mRNAs of the Transformer (Tra) gene of Drosophila melanogaster undergo alternative splicing via the alternative acceptor site mode. The gene Tra encodes a protein that is expressed only in females. The primary transcript of this gene contains an intron with two possible acceptor sites. In males, the upstream acceptor site is used. This causes a longer version of exon 2 to be included in the processed transcript, including an early stop codon. The resulting mRNA encodes a truncated protein product that is inactive. Females produce the master sex determination protein Sex lethal (Sxl). The Sxl protein is a splicing repressor that binds to an ISS in the RNA of the Tra transcript near the upstream acceptor site, preventing U2AF protein from binding to the polypyrimidine tract. This prevents the use of this junction, shifting the spliceosome binding to the downstream acceptor site. Splicing at this point bypasses the stop codon, which is excised as part of the intron. The resulting mRNA encodes an active Tra protein, which itself is a regulator of alternative splicing of other sex-related genes (see dsx above).[1]
Exon definition: Fas receptor
[edit]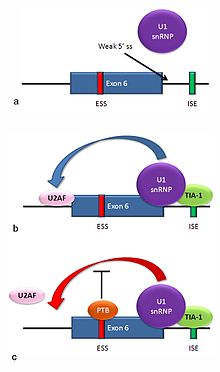
Multiple isoforms of the Fas receptor protein are produced by alternative splicing. Two normally occurring isoforms in humans are produced by an exon-skipping mechanism. An mRNA including exon 6 encodes the membrane-bound form of the Fas receptor, which promotes apoptosis, or programmed cell death. Increased expression of Fas receptor in skin cells chronically exposed to the sun, and absence of expression in skin cancer cells, suggests that this mechanism may be important in elimination of pre-cancerous cells in humans.[29] If exon 6 is skipped, the resulting mRNA encodes a soluble Fas protein that does not promote apoptosis. The inclusion or skipping of the exon depends on two antagonistic proteins, TIA-1 and polypyrimidine tract-binding protein (PTB).
- The 5' donor site in the intron downstream from exon 6 in the pre-mRNA has a weak agreement with the consensus sequence, and is not bound usually by the U1 snRNP. If U1 does not bind, the exon is skipped (see "a" in accompanying figure).
- Binding of TIA-1 protein to an intronic splicing enhancer site stabilizes binding of the U1 snRNP.[16] The resulting 5' donor site complex assists in binding of the splicing factor U2AF to the 3' splice site upstream of the exon, through a mechanism that is not yet known (see b).[30]
- Exon 6 contains a pyrimidine-rich exonic splicing silencer, ure6, where PTB can bind. If PTB binds, it inhibits the effect of the 5' donor complex on the binding of U2AF to the acceptor site, resulting in exon skipping (see c).
This mechanism is an example of exon definition in splicing. A spliceosome assembles on an intron, and the snRNP subunits fold the RNA so that the 5' and 3' ends of the intron are joined. However, recently studied examples such as this one show that there are also interactions between the ends of the exon. In this particular case, these exon definition interactions are necessary to allow the binding of core splicing factors prior to assembly of the spliceosomes on the two flanking introns.[30]
Repressor-activator competition: HIV-1 tat exon 2
[edit]
HIV, the retrovirus that causes AIDS in humans, produces a single primary RNA transcript, which is alternatively spliced in multiple ways to produce over 40 different mRNAs.[31] Equilibrium among differentially spliced transcripts provides multiple mRNAs encoding different products that are required for viral multiplication.[32] One of the differentially spliced transcripts contains the tat gene, in which exon 2 is a cassette exon that may be skipped or included. The inclusion of tat exon 2 in the RNA is regulated by competition between the splicing repressor hnRNP A1 and the SR protein SC35. Within exon 2 an exonic splicing silencer sequence (ESS) and an exonic splicing enhancer sequence (ESE) overlap. If A1 repressor protein binds to the ESS, it initiates cooperative binding of multiple A1 molecules, extending into the 5' donor site upstream of exon 2 and preventing the binding of the core splicing factor U2AF35 to the polypyrimidine tract. If SC35 binds to the ESE, it prevents A1 binding and maintains the 5' donor site in an accessible state for assembly of the spliceosome. Competition between the activator and repressor ensures that both mRNA types (with and without exon 2) are produced.[31]
Adaptive significance
[edit]Genuine alternative splicing occurs in both protein-coding genes and non-coding genes to produce multiple products (proteins or non-coding RNAs). External information is needed in order to decide which product is made, given a DNA sequence and the initial transcript. Since the methods of regulation are inherited, this provides novel ways for mutations to affect gene expression.[33]
Alternative splicing may provide evolutionary flexibility. A single point mutation may cause a given exon to be occasionally excluded or included from a transcript during splicing, allowing production of a new protein isoform without loss of the original protein.[1] Studies have identified intrinsically disordered regions (see Intrinsically unstructured proteins) as enriched in the non-constitutive exons[34] suggesting that protein isoforms may display functional diversity due to the alteration of functional modules within these regions. Such functional diversity achieved by isoforms is reflected by their expression patterns and can be predicted by machine learning approaches.[35][36] Comparative studies indicate that alternative splicing preceded multicellularity in evolution, and suggest that this mechanism might have been co-opted to assist in the development of multicellular organisms.[37]
Research based on the Human Genome Project and other genome sequencing has shown that humans have only about 30% more genes than the roundworm Caenorhabditis elegans, and only about twice as many as the fly Drosophila melanogaster. This finding led to speculation that the perceived greater complexity of humans, or vertebrates generally, might be due to higher rates of alternative splicing in humans than are found in invertebrates.[38][39] However, a study on samples of 100,000 expressed sequence tags (EST) each from human, mouse, rat, cow, fly (D. melanogaster), worm (C. elegans), and the plant Arabidopsis thaliana found no large differences in frequency of alternatively spliced genes among humans and any of the other animals tested.[40] Another study, however, proposed that these results were an artifact of the different numbers of ESTs available for the various organisms. When they compared alternative splicing frequencies in random subsets of genes from each organism, the authors concluded that vertebrates do have higher rates of alternative splicing than invertebrates.[41]
Disease
[edit]Changes in the RNA processing machinery may lead to mis-splicing of multiple transcripts, while single-nucleotide alterations in splice sites or cis-acting splicing regulatory sites may lead to differences in splicing of a single gene, and thus in the mRNA produced from a mutant gene's transcripts. A study in 2005 involving probabilistic analyses indicated that greater than 60% of human disease-causing mutations affect splicing rather than directly affecting coding sequences.[42] A more recent study indicates that one-third of all hereditary diseases are likely to have a splicing component.[20] Regardless of exact percentage, a number of splicing-related diseases do exist.[43] As described below, a prominent example of splicing-related diseases is cancer.
Abnormally spliced mRNAs are also found in a high proportion of cancerous cells.[44][45][46] Combined RNA-Seq and proteomics analyses have revealed striking differential expression of splice isoforms of key proteins in important cancer pathways.[47] It is not always clear whether such aberrant patterns of splicing contribute to the cancerous growth, or are merely consequence of cellular abnormalities associated with cancer. For certain types of cancer, like in colorectal and prostate, the number of splicing errors per cancer has been shown to vary greatly between individual cancers, a phenomenon referred to as transcriptome instability.[48][49] Transcriptome instability has further been shown to correlate grealty with reduced expression level of splicing factor genes. Mutation of DNMT3A has been demonstrated to contribute to hematologic malignancies, and that DNMT3A-mutated cell lines exhibit transcriptome instability as compared to their isogenic wildtype counterparts.[50]
In fact, there is actually a reduction of alternative splicing in cancerous cells compared to normal ones, and the types of splicing differ; for instance, cancerous cells show higher levels of intron retention than normal cells, but lower levels of exon skipping.[51] Some of the differences in splicing in cancerous cells may be due to the high frequency of somatic mutations in splicing factor genes,[46] and some may result from changes in phosphorylation of trans-acting splicing factors.[33] Others may be produced by changes in the relative amounts of splicing factors produced; for instance, breast cancer cells have been shown to have increased levels of the splicing factor SF2/ASF.[52] One study found that a relatively small percentage (383 out of over 26000) of alternative splicing variants were significantly higher in frequency in tumor cells than normal cells, suggesting that there is a limited set of genes which, when mis-spliced, contribute to tumor development.[53] It is believed however that the deleterious effects of mis-spliced transcripts are usually safeguarded and eliminated by a cellular posttranscriptional quality control mechanism termed nonsense-mediated mRNA decay [NMD].[54]
One example of a specific splicing variant associated with cancers is in one of the human DNMT genes. Three DNMT genes encode enzymes that add methyl groups to DNA, a modification that often has regulatory effects. Several abnormally spliced DNMT3B mRNAs are found in tumors and cancer cell lines. In two separate studies, expression of two of these abnormally spliced mRNAs in mammalian cells caused changes in the DNA methylation patterns in those cells. Cells with one of the abnormal mRNAs also grew twice as fast as control cells, indicating a direct contribution to tumor development by this product.[33]
Another example is the Ron (MST1R) proto-oncogene. An important property of cancerous cells is their ability to move and invade normal tissue. Production of an abnormally spliced transcript of Ron has been found to be associated with increased levels of the SF2/ASF in breast cancer cells. The abnormal isoform of the Ron protein encoded by this mRNA leads to cell motility.[52]
Overexpression of a truncated splice variant of the FOSB gene – ΔFosB – in a specific population of neurons in the nucleus accumbens has been identified as the causal mechanism involved in the induction and maintenance of an addiction to drugs and natural rewards.[55][56][57][58]
Recent provocative studies point to a key function of chromatin structure and histone modifications in alternative splicing regulation. These insights suggest that epigenetic regulation determines not only what parts of the genome are expressed but also how they are spliced.[59]
Genome-scale (transcriptome-wide) analysis
[edit]Transcriptome-wide analysis of alternative splicing is typically performed by high-throughput RNA-sequencing. Most commonly, by short-read sequencing, such as by Illumina instrumentation. But even more informative, by long-read sequencing, such as by Nanopore or PacBio instrumentation. Transcriptome-wide analyses can for example be used to measure the amount of deviating alternative splicing, such as in a cancer cohort.[60]
Deep sequencing technologies have been used to conduct genome-wide analyses of both unprocessed and processed mRNAs; thus providing insights into alternative splicing. For example, results from use of deep sequencing indicate that, in humans, an estimated 95% of transcripts from multiexon genes undergo alternative splicing, with a number of pre-mRNA transcripts spliced in a tissue-specific manner.[2] Functional genomics and computational approaches based on multiple instance learning have also been developed to integrate RNA-seq data to predict functions for alternatively spliced isoforms.[36] Deep sequencing has also aided in the in vivo detection of the transient lariats that are released during splicing, the determination of branch site sequences, and the large-scale mapping of branchpoints in human pre-mRNA transcripts.[61]
More historically, alternatively spliced transcripts have been found by comparing EST sequences, but this requires sequencing of very large numbers of ESTs. Most EST libraries come from a very limited number of tissues, so tissue-specific splice variants are likely to be missed in any case. High-throughput approaches to investigate splicing have, however, been developed, such as: DNA microarray-based analyses, RNA-binding assays, and deep sequencing. These methods can be used to screen for polymorphisms or mutations in or around splicing elements that affect protein binding. When combined with splicing assays, including in vivo reporter gene assays, the functional effects of polymorphisms or mutations on the splicing of pre-mRNA transcripts can then be analyzed.[20][23][62]
In microarray analysis, arrays of DNA fragments representing individual exons (e.g. Affymetrix exon microarray) or exon/exon boundaries (e.g. arrays from ExonHit or Jivan) have been used. The array is then probed with labeled cDNA from tissues of interest. The probe cDNAs bind to DNA from the exons that are included in mRNAs in their tissue of origin, or to DNA from the boundary where two exons have been joined. This can reveal the presence of particular alternatively spliced mRNAs.[63]
CLIP (Cross-linking and immunoprecipitation) uses UV radiation to link proteins to RNA molecules in a tissue during splicing. A trans-acting splicing regulatory protein of interest is then precipitated using specific antibodies. When the RNA attached to that protein is isolated and cloned, it reveals the target sequences for that protein.[64] Another method for identifying RNA-binding proteins and mapping their binding to pre-mRNA transcripts is "Microarray Evaluation of Genomic Aptamers by shift (MEGAshift)".net[65] This method involves an adaptation of the "Systematic Evolution of Ligands by Exponential Enrichment (SELEX)" method[66] together with a microarray-based readout. Use of the MEGAshift method has provided insights into the regulation of alternative splicing by allowing for the identification of sequences in pre-mRNA transcripts surrounding alternatively spliced exons that mediate binding to different splicing factors, such as ASF/SF2 and PTB.[67] This approach has also been used to aid in determining the relationship between RNA secondary structure and the binding of splicing factors.[22]
Use of reporter assays makes it possible to find the splicing proteins involved in a specific alternative splicing event by constructing reporter genes that will express one of two different fluorescent proteins depending on the splicing reaction that occurs. This method has been used to isolate mutants affecting splicing and thus to identify novel splicing regulatory proteins inactivated in those mutants.[64]
Recent advancements in protein structure prediction have facilitated the development of new tools for genome annotation and alternative splicing anlaysis. For instance, isoform.io, a platform guided by protein structure predictions, has evaluated hundreds of thousands of isoforms of human protein-coding genes assembled from numerous RNA sequencing experiments across a variety of human tissues. This comprehensive analysis has led to the identification of numerous isoforms with more confidently predicted structure and potentially superior function compared to canonical isoforms in the latest human gene database. By integrating structural predictions with expression and evolutionary evidence, this approach has demonstrated the potential of protein structure prediction as a tool for refining the annotation of the human genome.[68]
Databases
[edit]There is a collection of alternative splicing databases.[69][70][71] These databases are useful for finding genes having pre-mRNAs undergoing alternative splicing and alternative splicing events or to study the functional impact of alternative splicing.
- AspicDB database
- Intronerator database
- ProSAS database
See also
[edit]References
[edit]- ^ Jump up to: a b c d e f g h Black DL (2003). "Mechanisms of alternative pre-messenger RNA splicing" (PDF). Annual Review of Biochemistry. 72 (1): 291–336. doi:10.1146/annurev.biochem.72.121801.161720. PMID 12626338. S2CID 23576288.
- ^ Jump up to: a b c Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ (December 2008). "Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing". Nature Genetics. 40 (12): 1413–5. doi:10.1038/ng.259. PMID 18978789. S2CID 9228930.
- ^ Bhuiyan SA, Ly S, Phan M, Huntington B, Hogan E, Liu CC, Liu J, Pavlidis P (2018). "Systematic evaluation of isoform function in literature reports of alternative splicing". BMC Genomics. 19 (1): 637. doi:10.1186/s12864-018-5013-2. PMC 6114036. PMID 30153812. S2CID 52113302.
- ^ Tress ML, Abascal F, Valencia A (2017). "Alternative splicing may not be the key to proteome complexity". Trends in Biochemical Sciences. 42 (2): 98–110. doi:10.1016/j.tibs.2016.08.008. PMC 6526280. PMID 27712956.
- ^ Chow LT, Gelinas RE, Broker TR, Roberts RJ (September 1977). "An amazing sequence arrangement at the 5' ends of adenovirus 2 messenger RNA". Cell. 12 (1): 1–8. doi:10.1016/0092-8674(77)90180-5. PMID 902310. S2CID 2099968.
- ^ Berget SM, Moore C, Sharp PA (August 1977). "Spliced segments at the 5' terminus of adenovirus 2 late mRNA". Proceedings of the National Academy of Sciences of the United States of America. 74 (8): 3171–5. Bibcode:1977PNAS...74.3171B. doi:10.1073/pnas.74.8.3171. PMC 431482. PMID 269380.
- ^ Jump up to: a b c Leff SE, Rosenfeld MG, Evans RM (1986). "Complex transcriptional units: diversity in gene expression by alternative RNA processing". Annual Review of Biochemistry. 55 (1): 1091–117. doi:10.1146/annurev.bi.55.070186.005303. PMID 3017190.
- ^ Chow LT, Broker TR (October 1978). "The spliced structures of adenovirus 2 fiber message and the other late mRNAs". Cell. 15 (2): 497–510. doi:10.1016/0092-8674(78)90019-3. PMID 719751. S2CID 44642349.
- ^ Nevins JR, Darnell JE (December 1978). "Steps in the processing of Ad2 mRNA: poly(A)+ nuclear sequences are conserved and poly(A) addition precedes splicing". Cell. 15 (4): 1477–93. doi:10.1016/0092-8674(78)90071-5. PMID 729004. S2CID 39704416.
- ^ Rosenfeld MG, Amara SG, Roos BA, Ong ES, Evans RM (March 1981). "Altered expression of the calcitonin gene associated with RNA polymorphism". Nature. 290 (5801): 63–5. Bibcode:1981Natur.290...63R. doi:10.1038/290063a0. PMID 7207587. S2CID 4318349.
- ^ Rosenfeld MG, Lin CR, Amara SG, Stolarsky L, Roos BA, Ong ES, Evans RM (March 1982). "Calcitonin mRNA polymorphism: peptide switching associated with alternative RNA splicing events". Proceedings of the National Academy of Sciences of the United States of America. 79 (6): 1717–21. Bibcode:1982PNAS...79.1717R. doi:10.1073/pnas.79.6.1717. PMC 346051. PMID 6952224.
- ^ Maki R, Roeder W, Traunecker A, Sidman C, Wabl M, Raschke W, Tonegawa S (May 1981). "The role of DNA rearrangement and alternative RNA processing in the expression of immunoglobulin delta genes". Cell. 24 (2): 353–65. doi:10.1016/0092-8674(81)90325-1. PMID 6786756. S2CID 13208589.
- ^ Schmucker D, Clemens JC, Shu H, Worby CA, Xiao J, Muda M, et al. (June 2000). "Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity". Cell. 101 (6): 671–84. doi:10.1016/S0092-8674(00)80878-8. PMID 10892653. S2CID 13829976.
- ^ Jump up to: a b Westergren Jakobsson A, Segerman B, Wallerman O, Lind SB, Zhao H, Rubin CJ, et al. (November 2020). "The Human Adenovirus Type 2 Transcriptome: An Amazing Complexity of Alternatively Spliced mRNAs". Journal of Virology. 95 (4). doi:10.1128/JVI.01869-20. PMC 7851563. PMID 33239457.
- ^ Jump up to: a b c d e Sammeth M, Foissac S, Guigó R (August 2008). Brent MR (ed.). "A general definition and nomenclature for alternative splicing events". PLOS Computational Biology. 4 (8): e1000147. Bibcode:2008PLSCB...4E0147S. doi:10.1371/journal.pcbi.1000147. PMC 2467475. PMID 18688268.
- ^ Jump up to: a b c d e f g h Matlin AJ, Clark F, Smith CW (May 2005). "Understanding alternative splicing: towards a cellular code". Nature Reviews. Molecular Cell Biology. 6 (5): 386–98. doi:10.1038/nrm1645. PMID 15956978. S2CID 14883495.
- ^ Ner-Gaon, Hadas; Halachmi, Ronit; Savaldi-Goldstein, Sigal; Rubin, Eitan; Ophir, Ron; Fluhr, Robert (2004). "Intron retention is a major phenomenon in alternative splicing in Arabidopsis". The Plant Journal. 39 (6): 877–885. doi:10.1111/j.1365-313X.2004.02172.x. ISSN 1365-313X. PMID 15341630.
- ^ Gao K, Masuda A, Matsuura T, Ohno K (April 2008). "Human branch point consensus sequence is yUnAy". Nucleic Acids Research. 36 (7): 2257–67. doi:10.1093/nar/gkn073. PMC 2367711. PMID 18285363.
- ^ Clark D (2005). Molecular biology. Amsterdam: Elsevier Academic Press. ISBN 978-0-12-175551-5.
- ^ Jump up to: a b c Lim KH, Ferraris L, Filloux ME, Raphael BJ, Fairbrother WG (July 2011). "Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes". Proceedings of the National Academy of Sciences of the United States of America. 108 (27): 11093–8. Bibcode:2011PNAS..10811093H. doi:10.1073/pnas.1101135108. PMC 3131313. PMID 21685335.
- ^ Warf MB, Berglund JA (March 2010). "Role of RNA structure in regulating pre-mRNA splicing". Trends in Biochemical Sciences. 35 (3): 169–78. doi:10.1016/j.tibs.2009.10.004. PMC 2834840. PMID 19959365.
- ^ Jump up to: a b Reid DC, Chang BL, Gunderson SI, Alpert L, Thompson WA, Fairbrother WG (December 2009). "Next-generation SELEX identifies sequence and structural determinants of splicing factor binding in human pre-mRNA sequence". RNA. 15 (12): 2385–97. doi:10.1261/rna.1821809. PMC 2779669. PMID 19861426.
- ^ Jump up to: a b c d Wang Z, Burge CB (May 2008). "Splicing regulation: from a parts list of regulatory elements to an integrated splicing code" (Free full text). RNA. 14 (5): 802–13. doi:10.1261/rna.876308. PMC 2327353. PMID 18369186.
- ^ Jump up to: a b Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, et al. (May 2010). "Deciphering the splicing code". Nature. 465 (7294): 53–9. Bibcode:2010Natur.465...53B. doi:10.1038/nature09000. PMID 20445623. S2CID 2398858.
- ^ Ke S, Zhang XH, Chasin LA (April 2008). "Positive selection acting on splicing motifs reflects compensatory evolution". Genome Research. 18 (4): 533–43. doi:10.1101/gr.070268.107. PMC 2279241. PMID 18204002.
- ^ Fairbrother WG, Holste D, Burge CB, Sharp PA (September 2004). "Single nucleotide polymorphism-based validation of exonic splicing enhancers". PLOS Biology. 2 (9): E268. doi:10.1371/journal.pbio.0020268. PMC 514884. PMID 15340491.
- ^ Jump up to: a b Lynch KW, Maniatis T (August 1996). "Assembly of specific SR protein complexes on distinct regulatory elements of the Drosophila doublesex splicing enhancer". Genes & Development. 10 (16): 2089–101. doi:10.1101/gad.10.16.2089. PMID 8769651.
- ^ Graveley BR, Hertel KJ, Maniatis T (June 2001). "The role of U2AF35 and U2AF65 in enhancer-dependent splicing". RNA. 7 (6): 806–18. doi:10.1017/S1355838201010317. PMC 1370132. PMID 11421359.
- ^ Filipowicz E, Adegboyega P, Sanchez RL, Gatalica Z (February 2002). "Expression of CD95 (Fas) in sun-exposed human skin and cutaneous carcinomas". Cancer. 94 (3): 814–9. doi:10.1002/cncr.10277. PMID 11857317. S2CID 23772719.
- ^ Jump up to: a b Izquierdo JM, Majós N, Bonnal S, Martínez C, Castelo R, Guigó R, et al. (August 2005). "Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition". Molecular Cell. 19 (4): 475–84. doi:10.1016/j.molcel.2005.06.015. PMID 16109372.
- ^ Jump up to: a b Zahler AM, Damgaard CK, Kjems J, Caputi M (March 2004). "SC35 and heterogeneous nuclear ribonucleoprotein A/B proteins bind to a juxtaposed exonic splicing enhancer/exonic splicing silencer element to regulate HIV-1 tat exon 2 splicing". The Journal of Biological Chemistry. 279 (11): 10077–84. doi:10.1074/jbc.M312743200. PMID 14703516.
- ^ Jacquenet S, Méreau A, Bilodeau PS, Damier L, Stoltzfus CM, Branlant C (November 2001). "A second exon splicing silencer within human immunodeficiency virus type 1 tat exon 2 represses splicing of Tat mRNA and binds protein hnRNP H". The Journal of Biological Chemistry. 276 (44): 40464–75. doi:10.1074/jbc.M104070200. PMID 11526107.
- ^ Jump up to: a b c Fackenthal JD, Godley LA (2008). "Aberrant RNA splicing and its functional consequences in cancer cells" (Free full text). Disease Models & Mechanisms. 1 (1): 37–42. doi:10.1242/dmm.000331. PMC 2561970. PMID 19048051.
- ^ Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, Oldfield CJ, et al. (May 2006). "Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms". Proceedings of the National Academy of Sciences of the United States of America. 103 (22): 8390–5. Bibcode:2006PNAS..103.8390R. doi:10.1073/pnas.0507916103. PMC 1482503. PMID 16717195.
- ^ Li HD, Menon R, Omenn GS, Guan Y (August 2014). "The emerging era of genomic data integration for analyzing splice isoform function". Trends in Genetics. 30 (8): 340–7. doi:10.1016/j.tig.2014.05.005. PMC 4112133. PMID 24951248.
- ^ Jump up to: a b Eksi R, Li HD, Menon R, Wen Y, Omenn GS, Kretzler M, Guan Y (Nov 2013). "Systematically differentiating functions for alternatively spliced isoforms through integrating RNA-seq data". PLOS Computational Biology. 9 (11): e1003314. Bibcode:2013PLSCB...9E3314E. doi:10.1371/journal.pcbi.1003314. PMC 3820534. PMID 24244129.
- ^ Irimia M, Rukov JL, Penny D, Roy SW (October 2007). "Functional and evolutionary analysis of alternatively spliced genes is consistent with an early eukaryotic origin of alternative splicing". BMC Evolutionary Biology. 7 (1): 188. Bibcode:2007BMCEE...7..188I. doi:10.1186/1471-2148-7-188. PMC 2082043. PMID 17916237.
- ^ Ewing B, Green P (June 2000). "Analysis of expressed sequence tags indicates 35,000 human genes". Nature Genetics. 25 (2): 232–4. doi:10.1038/76115. PMID 10835644. S2CID 19165121.
- ^ Roest Crollius H, Jaillon O, Bernot A, Dasilva C, Bouneau L, Fischer C, et al. (June 2000). "Estimate of human gene number provided by genome-wide analysis using Tetraodon nigroviridis DNA sequence". Nature Genetics. 25 (2): 235–8. doi:10.1038/76118. PMID 10835645. S2CID 44052050.
- ^ Brett D, Pospisil H, Valcárcel J, Reich J, Bork P (January 2002). "Alternative splicing and genome complexity". Nature Genetics. 30 (1): 29–30. doi:10.1038/ng803. PMID 11743582. S2CID 2724843.
- ^ Kim E, Magen A, Ast G (2006). "Different levels of alternative splicing among eukaryotes". Nucleic Acids Research. 35 (1): 125–31. doi:10.1093/nar/gkl924. PMC 1802581. PMID 17158149.
- ^ López-Bigas N, Audit B, Ouzounis C, Parra G, Guigó R (March 2005). "Are splicing mutations the most frequent cause of hereditary disease?". FEBS Letters. 579 (9): 1900–3. doi:10.1016/j.febslet.2005.02.047. PMID 15792793. S2CID 30174458.
- ^ Ward AJ, Cooper TA (January 2010). "The pathobiology of splicing". The Journal of Pathology. 220 (2): 152–63. doi:10.1002/path.2649. PMC 2855871. PMID 19918805.
- ^ Skotheim RI, Nees M (2007). "Alternative splicing in cancer: noise, functional, or systematic?". The International Journal of Biochemistry & Cell Biology. 39 (7–8): 1432–49. doi:10.1016/j.biocel.2007.02.016. PMID 17416541.
- ^ He C, Zhou F, Zuo Z, Cheng H, Zhou R (2009). Bauer JA (ed.). "A global view of cancer-specific transcript variants by subtractive transcriptome-wide analysis". PLOS ONE. 4 (3): e4732. Bibcode:2009PLoSO...4.4732H. doi:10.1371/journal.pone.0004732. PMC 2648985. PMID 19266097.
- ^ Jump up to: a b Sveen A, Kilpinen S, Ruusulehto A, Lothe RA, Skotheim RI (May 2016). "Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes". Oncogene. 35 (19): 2413–27. doi:10.1038/onc.2015.318. PMID 26300000. S2CID 22943729.
- ^ Omenn GS, Guan Y, Menon R (July 2014). "A new class of protein cancer biomarker candidates: differentially expressed splice variants of ERBB2 (HER2/neu) and ERBB1 (EGFR) in breast cancer cell lines". Journal of Proteomics. 107: 103–12. doi:10.1016/j.jprot.2014.04.012. PMC 4123867. PMID 24802673.
- ^ Sveen A, Johannessen B, Teixeira MR, Lothe RA, Skotheim RI (August 2014). "Transcriptome instability as a molecular pan-cancer characteristic of carcinomas". BMC Genomics. 15 (1): 672. doi:10.1186/1471-2164-15-672. PMC 3219073. PMID 25109687.
- ^ Sveen A, Agesen TH, Nesbakken A, Rognum to, Lothe RA, Skotheim RI (май 2011 г.). «Транскриптом нестабильность при колоректальном раке, идентифицированная с помощью анализа микрочипов экзонов: ассоциации с уровнями экспрессии фактора сплайсинга и выживаемостью пациента» . Медицина генома . 3 (5): 32. doi : 10.1186/gm248 . PMC 4137096 . PMID 21619627 .
- ^ Banaszak LG, Giudice V, Zhao X, Wu Z, Gao S, Hosokawa K, et al. (Март 2018 г.). «Аномальное сплайсинг РНК и нестабильность генома после индукции мутаций DNMT3A с помощью редактирования генов CRISPR/CAS9» . Клетки крови, молекулы и заболевания . 69 : 10–22. doi : 10.1016/j.bcmd.2017.12.002 . PMC 6728079 . PMID 29324392 .
- ^ Ким Э, Горен А., Аст Г (январь 2008 г.). «Понимает связь между раком и альтернативным сплайсингом». Тенденции в генетике . 24 (1): 7–10. doi : 10.1016/j.tig.2007.10.001 . PMID 18054115 .
- ^ Jump up to: а беременный Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM, et al. (Декабрь 2005 г.). «Моторичность клеток контролируется SF2/ASF посредством альтернативного сплайсинга Ron Protooncogene» . Молекулярная клетка . 20 (6): 881–90. doi : 10.1016/j.molcel.2005.10.026 . PMID 16364913 .
- ^ Hui L, Zhang X, Wu X, Lin Z, Wang Q, Li Y, Hu G (апрель 2004 г.). «Идентификация альтернативно сплайсированных вариантов мРНК, связанных с раковым раком с помощью выравнивания EST по всему геному» . Онкоген . 23 (17): 3013–23. doi : 10.1038/sj.onc.1207362 . PMID 15048092 .
- ^ Danckwardt S, Neu-Yilik G, Thermann R, Frede U, Hentze MW, Kulozik AE (март 2002 г.). «Аномально сплайсированные мРНК бета-глобина: единственная точечная мутация генерирует транскрипты чувствительными и нечувствительными к распаданию мРНК, опосредованной бессмысленностью» . Кровь . 99 (5): 1811–6. doi : 10.1182/blood.v99.5.1811 . PMID 11861299 . S2CID 17128174 .
- ^ Nestler EJ (декабрь 2013 г.). «Сотовая основа памяти для зависимости» . Диалоги в клинической нейробиологии . 15 (4): 431–43. doi : 10.31887/dcns.2013.15.4/enestler . PMC 3898681 . PMID 24459410 .
Несмотря на важность многочисленных психосоциальных факторов, в своей основе наркомания включает в себя биологический процесс: способность повторного воздействия лекарства от злоупотреблений вызывать изменения в уязвимом мозге, который стимулирует навязчивое поиск и принятие лекарств, и потеря контроля За употреблением наркотиков это определяет состояние зависимости. ... Большая литература продемонстрировала, что такая индукция ΔFOSB в нейронах NAC типа D1 повышает чувствительность животного к лекарственным средствам, а также естественные награды и способствует самостоятельному применению лекарств, предположительно в процессе положительного подкрепления
- ^ Ruffle JK (ноябрь 2014). «Молекулярная нейробиология зависимости: о чем все (δ) FOSB?». Американский журнал по вопросам злоупотребления наркотиками и алкоголем . 40 (6): 428–37. doi : 10.3109/00952990.2014.933840 . PMID 25083822 . S2CID 19157711 .
ΔFOSB является важным транскрипционным фактором, участвующим в молекулярных и поведенческих путях зависимости после повторного воздействия лекарственного средства. Образование ΔFOSB в нескольких областях мозга, и молекулярный путь, ведущий к образованию комплексов AP-1, хорошо понятно. Создание функциональной цели для ΔFOSB позволило дополнительно определить некоторые из ключевых аспектов его молекулярных каскадов, включающих такие эффекторы, как GluR2 (87,88), CDK5 (93) и NFKB (100). Более того, многие из этих идентифицированных молекулярных изменений в настоящее время напрямую связаны с структурными, физиологическими и поведенческими изменениями, наблюдаемыми после хронического воздействия лекарственного средства (60,95,97,102). Новые границы исследований, изучающих молекулярные роли ΔFOSB, были открыты эпигенетическими исследованиями, и последние достижения иллюстрировали роль ΔFOSB, действующего на ДНК и гистоны, действительно как Молекулярный переключатель (34). Как следствие нашего улучшенного понимания ΔFOSB в зависимости, можно оценить привыкающий потенциал современных лекарств (119), а также использовать его в качестве биомаркера для оценки эффективности терапевтических вмешательств (121,122,124). Некоторые из этих предлагаемых вмешательств имеют ограничения (125) или находятся в их младенчестве (75). Тем не менее, есть надежда, что некоторые из этих предварительных результатов могут привести к инновационным методам лечения, которые очень необходимы в зависимости.
- ^ Biliński P, Wojtył A, Kapka-Skrzypczak L, Chwedorowicz R, Cyranka M, Studziński T (2012). «Эпигенетическая регуляция в наркомании». Анналы сельскохозяйственной и экологической медицины . 19 (3): 491–6. PMID 23020045 .
По этим причинам ΔFOSB считается первичным и причинным фактором транскрипции при создании новых нейронных связей в центре вознаграждения, префронтальной коре и других областях лимбической системы. Это отражается на повышенном, стабильном и долгосрочном уровне чувствительности к кокаину и другим лекарствам, а также склонности к рецидиве даже после длительных периодов воздержания. Эти вновь построенные сети функционируют очень эффективно через новые пути, как только принимаются наркотики, злоупотребление
- ^ Olsen CM (декабрь 2011 г.). «Естественные награды, нейропластичность и не лекарства» . Нейрофармакология . 61 (7): 1109–22. doi : 10.1016/j.neuropharm.2011.03.010 . PMC 3139704 . PMID 21459101 .
- ^ Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T (январь 2011 г.). «Эпигенетика в альтернативном сплайсингах до мРНК» . Клетка . 144 (1): 16–26. doi : 10.1016/j.cell.2010.11.056 . PMC 3038581 . PMID 21215366 .
- ^ Strømme JM, Johannessen B, Skotheim Ri (2023). «Определение альтернативного сплайсинга в качестве молекулярного подтипа микросателлитного стабильного колоректального рака» . JCO Клинический рак информатики . 7 (7): E2200159. doi : 10.1200/cci.22.00159 . HDL : 10852/108838 . PMID 36821799 .
- ^ Taggart AJ, Desimone AM, Shih JS, Filloux ME, Fairbrother WG (июнь 2012 г.). «Крупномасштабное картирование топок ветвей в транскриптах до-мРНК человека in vivo» . Природа структурная и молекулярная биология . 19 (7): 719–21. doi : 10.1038/nsmb.2327 . PMC 3465671 . PMID 22705790 .
- ^ Fairbrother WG, Yeh RF, Sharp PA, Burge CB (август 2002 г.). «Прогнозирующая идентификация экзонических усилителей сплайсинга в генах человека» . Наука . 297 (5583): 1007–13. Bibcode : 2002sci ... 297.1007f . doi : 10.1126/science.1073774 . PMID 12114529 . S2CID 8689111 .
- ^ Pan Q, Shai O, Misquitta C, Zhang W, Saltzman AL, Mohammad N, et al. (Декабрь 2004 г.). «Выявление глобальных регуляторных особенностей альтернативного сплайсинга млекопитающих с использованием количественной платформы микрочипов» . Молекулярная клетка . 16 (6): 929–41. doi : 10.1016/j.molcel.2004.12.004 . PMID 15610736 .
- ^ Jump up to: а беременный Дэвид С.Дж., Мэнли Дж.Л. (февраль 2008 г.). «Поиск альтернативных регуляторов сплайсинга: новые подходы предлагают путь к коду сплайсинга» . Гены и развитие . 22 (3): 279–85. doi : 10.1101/gad.1643108 . PMC 2731647 . PMID 18245441 .
- ^ Уоткинс К.Х., Стюарт А., Фэрбротер В. (декабрь 2009 г.). «Быстрый высокопроизводительный метод для картирования рибонуклеопротеинов (RNP) на пре-мРНК человека» . Журнал визуализированных экспериментов . 34 (34): 1622. doi : 10.3791/1622 . PMC 3152247 . PMID 19956082 .
- ^ Tuerk C, Gold L (август 1990 г.). «Систематическая эволюция лигандов путем экспоненциального обогащения: РНК -лиганды для бактериофага ДНК -полимеразы». Наука . 249 (4968): 505–10. Bibcode : 1990sci ... 249..505t . doi : 10.1126/science.2200121 . PMID 2200121 .
- ^ Чанг Б., Левин Дж., Томпсон В.А., Фэрбротер В.Г. (март 2010 г.). «Анализ высокопроизводительного связывания определяет специфичность связывания ASF/SF2 на альтернативно сплайсированной пре-мрНК человека» . Комбинаторная химия и высокая пропускная способность . 13 (3): 242–52. doi : 10.2174/138620710790980522 . PMC 3427726 . PMID 20015017 .
- ^ Соммер, Маркус Дж.; Ча, sooyoung; Varabyou, Ales; Ринкон, Наталья; Парк, Сукхван; Минкин, Илия; Pertea, Mihaela; Steinegger, Martin; Зальцберг, Стивен Л. (2022-12-15). «Структурная идентификация изоформы для человеческого транскриптома» . элиф . 11 : E82556. doi : 10.7554/elife.82556 . PMC 9812405 . PMID 36519529 .
- ^ Tapial J, Ha KC, Sterne-Weiler T, Gohr A, Braunschweig U, Hermoso-Pulido A, et al. (Октябрь 2017). «Атлас альтернативных профилей сплайсинга и функциональных ассоциаций выявляет новые регуляторные программы и гены, которые одновременно выражают несколько основных изоформ» . Исследование генома . 27 (10): 1759–1768. doi : 10.1101/gr.220962.117 . PMC 5630039 . PMID 28855263 .
- ^ Луади З., Юань К., Гресс А., Цой О., Калинина О.В., Баумбач Дж. И др. (Январь 2021 г.). «Digger: изучение функциональной роли альтернативного сплайсинга в белковых взаимодействиях» . Исследование нуклеиновых кислот . 49 (D1): D309 - D318. doi : 10.1093/nar/gkaa768 . PMC 7778957 . PMID 32976589 .
- ^ Родригес Дж. М., Майетта П., Эзкурдия И., Стритэлли А., Весселинк Дж.Дж., Лопес Г. и др. (Janogy 2013). «Утверждение: аннотация основных и альтернативных изоформ сплайсинга» . Исследование нуклеиновых кислот . 41 (база данных проблем): D110-7. Doi : 10.1093/nar/gks1058 . PMC 3531113 . PMID 23161672 .
Внешние ссылки
[ редактировать ]- Общее определение и номенклатура для альтернативных событий сплайсинга в Scivee
- Astalavista (альтернативный инструмент визуализации ландшафта сплайсинга), метод для вычислительной классификации альтернативных структур сплайсинга, архивных 2009-12-11 на машине Wayback
- Isopred: вычислительно прогнозируемые функции изоформ
- Stamms-lab.net: исследовательская группа, занимающаяся альтернативными проблемами сплайсинга и неправильным сбором при заболеваниях человека
- Альтернативный сплайсинг ионных каналов в мозге, связанный с психическими и неврологическими заболеваниями
- Bipass: веб -сервисы в альтернативном сплайсинге