Ферментный катализ
Ферментный катализ это увеличение скорости процесса с - помощью « фермента », биологической молекулы . Большинство ферментов являются белками, и большинство таких процессов являются химическими реакциями. Внутри фермента, как правило, катализ происходит на локализованном участке, называемом активным сайтом .
Большинство ферментов изготовлены преимущественно из белков, либо одной цепь белков, либо многих таких цепей в мульти-субъединичном комплексе . Ферменты часто также включают небелковые компоненты, такие как ионы металлов или специализированные органические молекулы, известные как кофактор (например, аденозин трихосфат ). Многие кофакторы являются витаминами, и их роль в качестве витаминов напрямую связана с их использованием в катализе биологического процесса в метаболизме. Катализ биохимических реакций в клетке жизненно важен, поскольку многие, но не все метаболически важные реакции имеют очень низкие скорости при некатируемом. Одним из факторов эволюции белка является оптимизация такой каталитической активности, хотя только самые важные ферменты работают вблизи лимитов каталитической эффективности, и многие ферменты далеки от оптимальных. Важные факторы в катализе фермента включают общий кислотный и базовый катализ , орбитальное рулевое управление, энтропийное ограничение, эффекты ориентации (то есть блокировка и ключевой катализ), а также эффекты движения, связанные с динамикой белка [ 1 ]
Механизмы ферментативного катализа различаются, но все в принципе схожи с другими типами химического катализа тем, что решающим фактором является снижение энергетического барьера, отделяющего реагенты (или субстраты ) от продуктов. Снижение энергии активации ( e a ) увеличивает долю молекул реагента, которые могут преодолеть этот барьер и сформировать продукт. Важным принципом является то, что, поскольку они уменьшают только энергетические барьеры между продуктами и реагентами, ферменты всегда катализируют реакции в обоих направлениях и не могут стимулировать реакцию вперед или влиять на положение равновесия - только скорость, с которой она достигается. Как и в других катализаторах, фермент не потребляется и не изменяется реакцией (как и субстрат), а переработано таким образом, что один фермент выполняет много раундов катализа.
Ферменты часто очень специфичны и действуют только на определенные субстраты. Некоторые ферменты являются абсолютно специфическими, что они действуют только на одном субстрате, в то время как другие демонстрируют специфичность группы и могут действовать на аналогичных, но не идентичных химических группах, таких как пептидная связь в разных молекулах. Многие ферменты имеют стереохимическую специфичность и действуют на одном стереоизомере , но не на другом. [ 2 ]
Индуцированная подгонка
[ редактировать ]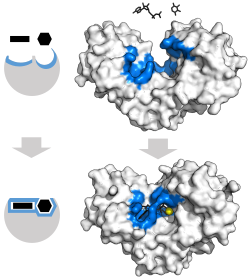
Классической моделью для взаимодействия фермента- субстрата является индуцированная модель FIT. [ 3 ] Эта модель предполагает, что начальное взаимодействие между ферментом и субстратом было относительно слабым, но что эти слабые взаимодействия быстро вызывают конформационные изменения в ферменте, который укрепляет связывание.
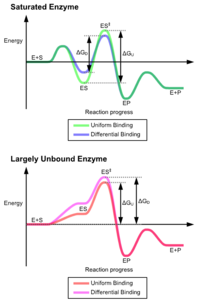
Преимущества индуцированного механизма соответствия возникают из -за стабилизирующего эффекта сильного связывания фермента. Существует два различных механизмах связывания субстрата: равномерное связывание, которое имеет сильное связывание субстрата и дифференциальное связывание, которое имеет сильное связывание переходного состояния. Стабилизирующий эффект равномерного связывания увеличивает аффинность как субстрата, так и переходного состояния, в то время как дифференциальное связывание увеличивает только аффинность связывания переходного состояния. Оба используются ферментами и были эволюционно выбраны для минимизации энергии активации реакции. Ферменты, которые насыщены, то есть имеют связывание подложки с высокой аффинностью, требуют дифференциального связывания, чтобы уменьшить энергию активации, тогда как небольшие несвязанные ферменты субстрата могут использовать либо дифференциальное, так и однородное связывание. [ 4 ]
Эти эффекты привели к тому, что большинство белков с использованием механизма дифференциального связывания для снижения энергии активации, поэтому большинство субстратов имеют высокую аффинность к ферменту во время переходного состояния. Дифференциальное связывание осуществляется с помощью индуцированного механизма соответствия - субстрат сначала связывается слабо, а затем фермент меняет конформацию, увеличивая аффинность к переходному состоянию и стабилизируя его, таким образом уменьшая энергию активации, чтобы достичь ее.
Однако важно уточнить, что концепция индуцированной соответствия не может быть использована для рационализации катализа. То есть химический катализ определяется как восстановление E A ‡ (Когда система уже находится в ES ‡ ) относительно E a ‡ в некатализированной реакции в воде (без фермента). Индуцированная подгонка только предполагает, что барьер ниже в закрытой форме фермента, но не говорит нам, в чем причина сокращения барьера.
Индуцированная соответствие может быть полезной для верности молекулярного распознавания в присутствии конкуренции и шума с помощью конформационного механизма корректуры . [ 5 ]
Механизмы альтернативного пути реакции
[ редактировать ]Эти конформационные изменения также приводят к каталитическим остаткам в активном участке, близком к химическим связям в субстрате, которые будут изменены в реакции. реакции После того, как связывание происходит, один или несколько механизмов катализа снижают энергию переходного состояния , предоставляя альтернативный химический путь для реакции. Существует шесть возможных механизмов катализа «над барьером», а также механизм «через барьер»:
Близость и ориентация
[ редактировать ]Взаимодействия фермента-субстрата выравнивают реактивные химические группы и держат их близко друг к другу в оптимальной геометрии, которая увеличивает скорость реакции. Это уменьшает энтропию реагентов и, таким образом, делает реакции добавления или переноса менее неблагоприятными, поскольку снижение общей энтропии, когда два реагента становятся одним продуктом. Однако это общий эффект и наблюдается в реакциях без приема или переноса, где он происходит из-за увеличения «эффективной концентрации» реагентов. Это понимается при рассмотрении того, как увеличение концентрации приводит к увеличению скорости реакции: по существу, когда реагенты более концентрируются, они сталкиваются чаще и поэтому реагируют чаще. В ферментативном катализе связывание реагентов с ферментом ограничивает конформационное пространство реагентов, удерживая их в «правильной ориентации» и близко друг к другу, чтобы они сталкивались чаще и с правильной геометрией, чтобы облегчить желаемая реакция. «Эффективная концентрация» - это концентрация, которой должен быть реагент, свободный в растворе, чтобы испытывать ту же столкновную частоту. Часто такие теоретические эффективные концентрации являются нефизическими и невозможно реализовать в реальности - что является свидетельством большой каталитической силы многих ферментов, с огромным увеличением скорости по сравнению с некатализированным состоянием.
| Например: |
| Подобные реакции будут происходить намного быстрее, если реакция является внутримолекулярной. |

|
| Эффективная концентрация ацетата во внутримолекулярной реакции может быть оценена как k 2 /k 1 = 2 x 10 5 Молярный. |
Тем не менее, ситуация может быть более сложной, поскольку современные вычислительные исследования установили, что традиционные примеры эффектов близости не могут быть непосредственно связаны с энтропическими эффектами фермента. [ 6 ] [ 7 ] [ 8 ] Также оригинальное энтропийное предложение [ 9 ] Было установлено, что в значительной степени переоценивает вклад энтропии ориентации в катализ. [ 10 ]
Доноры или акцепторы протонов
[ редактировать ]Доноры и акцепторы протонов, то есть кислоты и база могут пожертвовать и принимать протоны, чтобы стабилизировать развивающиеся расходы в переходном состоянии. Это связано с общим принципом катализа, принципа снижения энергетических барьеров, поскольку в целом переходные состояния являются высокими энергетическими состояниями, и путем стабилизации их этой высокой энергии снижается, снижая барьер. Ключевой особенностью ферментативного катализа во многих небиологических катализе является то, что как кислотный, так и базовый катализ может быть объединен в одной и той же реакции. Во многих абиотических системах кислоты (крупные [H+]) или основания (большая концентрация H+ раковины или виды с парами электронов) могут увеличить скорость реакции; Но, конечно, окружающая среда может иметь только один общий рН (мера кислотности или основности (щелочность)). Однако, поскольку ферменты представляют собой большие молекулы, они могут позиционировать как кислотные группы, так и основные группы в их активном участке для взаимодействия с их субстратами и использовать оба режима, независимые от объемного рН.
Часто общий кислотный или базовый катализ используется для активации нуклеофильных и/или электрофильных групп или для стабилизации уходящих групп. Многие аминокислоты с кислыми или основными группами используются в активном сайте, таких как глутамическая и аспарагиновая кислота, гистидин, цистин, тирозин, лизин и аргинин, а также серин и треонин. Кроме того, часто используется магистраль пептидов с карбонилом и амидными N -группами. Цистин и гистидин очень часто участвуют, поскольку они оба имеют PKA близко к нейтральному pH и поэтому могут принять и пожертвовать протоны.
Многие механизмы реакции, включающие кислотный/базовый катализ, предполагают существенно измененный PKA. Это изменение PKA возможно через местную среду остатка [ Цитация необходима ] .
| Условия | Кислоты | Базы |
|---|---|---|
| Гидрофобная среда | Увеличьте PKA | Уменьшить PKA |
| Соседние остатки аналогичного заряда | Увеличьте PKA | Уменьшить PKA |
| Соляный мост (и водород облигация) Формирование |
Уменьшить PKA | Увеличьте PKA |
На PKA также можно значительно влиять на окружающую среду, в той степени, в которой остатки, которые являются основными по решению, могут выступать в качестве доноров протонов, и наоборот.
| Например: |
| Каталитическая триада сериновой протеазы |

|
| Начальный этап каталитического механизма сериновой протеазы включает в себя гистидин активного участка, принимающего протон из остатка серина. Это готовит серин как нуклеофил, чтобы атаковать амидную связь субстрата. Этот механизм включает в себя пожертвование протона от серина (основание, PKA 14) до гистидина (кислота, PKA 6), ставшего возможным из -за локальной среды баз. |
Модификация PKA является чистой частью электростатического механизма. [ 11 ] Каталитический эффект приведенного выше примера в основном связан с восстановлением PKA оксианиона и увеличением PKA гистидина, в то время как перенос протона от серина к гистидина не катализируется значительно, поскольку это не скорость Определение барьера. [ 12 ] Обратите внимание, что в показанном примере гистидиновая конъюгатная кислота действует как общий кислотный катализатор для последующей потери амина из тетраэдрического промежутка. Доказательства, подтверждающие этот предложенный механизм (рис. 4 в реф. 13) [ 13 ] однако был соперным. [ 14 ]
Электростатический катализ
[ редактировать ]Стабилизация заряженных переходных состояний также может быть проведена остатками в активном участке, образующих ионные связи (или частичное взаимодействие ионного заряда) с промежуточным. Эти связи могут поступать либо из кислых , либо основных боковых цепей, обнаруженных на аминокислотах, таких как лизин , аргинин , аспартациновая кислота или глутаминовая кислота, металлов, или поступать из кофакторов таких как цинк . Ионы металлов особенно эффективны и могут уменьшить PKA воды достаточно, чтобы сделать его эффективным нуклеофилом.
Систематические исследования компьютерного моделирования установили, что электростатические эффекты дают, безусловно, самый большой вклад в катализ. [ 11 ] Это может увеличить скорость реакции в течение 10 до 10 7 . [ 15 ] В частности, было обнаружено, что фермент обеспечивает среду, которая является более полярной, чем вода, и что ионные переходные состояния стабилизируются фиксированными диполями. Это сильно отличается от стабилизации переходного состояния в воде, где молекулы воды должны платить с «энергией реорганизации». [ 16 ] Для стабилизации ионных и заряженных состояний. Таким образом, катализ связан с тем фактом, что ферментные полярные группы преорганизованы [ 17 ]
Было показано, что величина электростатического поля, проявляемое активным местом фермента, сильно коррелирует с повышением каталитической скорости фермента. [ 18 ]
Связывание субстрата обычно исключает воду из активного участка, тем самым снижая локальную диэлектрическую постоянную к кондиционеру органического растворителя. Это усиливает электростатические взаимодействия между заряженными/полярными субстратами и активными сайтами. Кроме того, исследования показали, что распределение зарядов по активным участкам расположены таким образом, чтобы стабилизировать переходные состояния катализируемых реакций. В нескольких ферментах эти распределения зарядов, по-видимому, служат для направления полярных субстратов к их сайтам связывания, так что скорости этих ферментативных реакций превышают их пределы, контролируемые диффузией [ Цитация необходима ] .
| Например: |
| карбоксипептидазы Каталитический механизм |

|
| Тетраэдрическое промежуточное соединение стабилизируется частичной ионной связью между Zn 2+ Ион и отрицательный заряд на кислороде. |
Ковалентный катализ
[ редактировать ]Ковалентный катализ включает субстрат, образующий переходную ковалентную связь с остатками в активном сайте фермента или с кофактором. Это добавляет дополнительное ковалентное промежуточное соединение реакции и помогает уменьшить энергию последующих переходных состояний реакции. Ковалентная связь должна на более поздней стадии реакции быть сломана, чтобы регенерировать фермент. Этот механизм используется каталитической триадой ферментов, таких как протеазы, такие как химотрипсин и трипсин , где образуется промежуточное здание ацил-анзиния. Альтернативный механизм - это образование основания SCHIFF с использованием свободного амина из остатка лизина , как видно из фермента альдолазы во время гликолиза .
В некоторых ферментах используются неаминокислотные кофакторы, такие как пиридоксальный фосфат (PLP) или тиамин пирофосфат (TPP), для образования ковалентных промежуточных соединений с молекулами реагента. [ 19 ] [ 20 ] Такие ковалентные промежуточные соединения функционируют, чтобы уменьшить энергию последующих переходных состояний, аналогично тому, как ковалентные промежуточные продукты, образованные с активным сайтом аминокислот, позволяют стабилизацию, но возможности кофакторов позволяют ферментам выполнять реакции, которые только аминокислотные остатки не могли. Ферменты, использующие такие кофакторы, включают PLP-зависимую ферментную аспартат-трансаминазу и TPP-зависимую фермент пируватдегидрогеназу . [ 21 ] [ 22 ]
Вместо того, чтобы снизить энергию активации для пути реакции, ковалентный катализ обеспечивает альтернативный путь для реакции (через ковалентный промежуточный), и поэтому отличается от истинного катализа. [ 11 ] Например, энергетику ковалентной связи с сериновой молекулой в химотрипсине следует сравнивать с хорошо понятой ковалентной связью с нуклеофилом в некватализированной реакции раствора. Истинное предложение о ковалентном катализе (где барьер ниже, чем соответствующий барьер в растворе), потребует, например, частичной ковалентной связи с переходным состоянием ферментной группой (например, очень прочная водородная связь) и тому подобное Эффекты не вносят значительный вклад в катализ.
Катализ ионов металлов
[ редактировать ]Ион металла в активном участке участвует в катализе путем координации стабилизации заряда и экранирования. Из -за положительного заряда металла только отрицательные заряды могут быть стабилизированы через ионы металлов. [ 23 ] Тем не менее, ионы металлов выгодны в биологическом катализе, потому что на них не влияют изменения в рН. [ 24 ] Ионы металлов также могут действовать, чтобы ионизировать воду, выступая в качестве кислоты Льюиса . [ 25 ] Ионы металлов также могут быть агентами окисления и восстановления. [ 26 ]
Напряжение связи
[ редактировать ]Это является основным эффектом индуцированного связывания FIT, где аффинность фермента к переходному состоянию больше, чем к самому субстрату. Это вызывает структурные перестройки, которые напрягают субстратные связи в положение ближе к конформации переходного состояния, таким образом снижая разницу в энергии между субстратом и переходным состоянием и помогая катализировать реакцию.
Однако эффект деформации, на самом деле, является эффектом дестабилизации основного состояния, а не эффектом стабилизации переходного состояния. [ 11 ] [ 27 ] [ страница необходима ] Кроме того, ферменты очень гибкие, и они не могут применить большой эффект деформации. [ 28 ]
В дополнение к деформации связей в субстрате, деформация связей также может быть индуцирован в самом ферменте для активации остатков в активном сайте.
| Например: |
| Субстрат, связанный субстрат и переходные состояния конформации лизоцима . |
| Субстрат при связывании искажена из конформации половины стула гексозного кольца (из -за стерического препятствия с аминокислотами белка, заставляющим экваториальный C6 находиться в осевой положении) к конформации стула,, [ 29 ] что по форме схожи с переходным состоянием. |
Квантовое туннелирование
[ редактировать ]Эти традиционные механизмы «над барьером» в некоторых случаях были оспаривались моделями и наблюдениями «через барьер» ( квантовое туннелирование ). Некоторые ферменты работают с кинетикой, которая быстрее, чем то, что было бы предсказано классическим Δg ‡ Полем В моделях «Через барьер» протон или электрон может туннель через барьеры активации. [ 30 ] [ 31 ] Квантовое туннелирование для протонов наблюдалось при триптамина окислении ароматической аминдегидрогеназой . [ 32 ]
Квантовое туннелирование, по -видимому, не обеспечивает серьезного каталитического преимущества, поскольку вклад туннелирования схож в катализируемых и некватизируемых реакциях в растворе. [ 31 ] [ 33 ] [ 34 ] [ 35 ] Однако вклад туннелирования (обычно повышение константы скорости в течение ~ 1000 [ 32 ] По сравнению со скоростью реакции для классического «над барьерным» маршрутом), вероятно, имеет решающее значение для жизнеспособности биологических организмов. Это подчеркивает общую важность туннельных реакций в биологии.
В 1971-1972 годах была сформулирована первая квантовомеханическая модель ферментного катализа. [ 36 ] [ 37 ] [ Требуется сторонний источник ]
Активный фермент
[ редактировать ]Энергия связывания комплекса фермента-субстрата не может рассматриваться как внешняя энергия, которая необходима для активации субстрата. Фермент высокоэнергетического содержания может сначала перенести некоторую специфическую энергетическую группу x 1 из каталитического сайта фермента на конечное место первого связанного реагента, а затем другая группа x 2 от второго граничного реагента (или из второй группы сингла Реагент) должен быть перенесен на активное участок для завершения преобразования субстрата в регенерацию продукта и фермента. [ 38 ]
Мы можем представить всю ферментативную реакцию в качестве двух реакций связи:
| S 1 + EX 1 → S 1 EX 1 → P 1 + EP 2 | ( 1 ) |
| S 2 + EP 2 → S 2 EP 2 → P 2 + EX 2 | ( 2 ) |
) можно увидеть Из реакции ( 1 , что группа x 1 активного фермента появляется в продукте из -за возможности реакции обмена внутри фермента, чтобы избежать как электростатического ингибирования, так и отталкивания атомов. Таким образом, мы представляем активный фермент как мощный реагент ферментативной реакции. Реакция ( 2 ) показывает неполное преобразование субстрата, поскольку его группа x 2 остается внутри фермента. Этот подход как идея ранее предполагал, полагаясь на гипотетические чрезвычайно высокие ферментативные конверсии (каталитически совершенный фермент). [ 39 ]
Важнейшим моментом для проверки настоящего подхода является то, что катализатор должен быть комплексом фермента с группой переноса реакции. Этот химический аспект подтверждается хорошо изученными механизмами нескольких ферментативных реакций. Рассмотрим реакцию гидролиза пептидной связи, катализируемой чистым белком α-химотрипсином (фермент, действующий без кофактора), который является хорошо изученным членом семейства сериновых протеасов, см. [ 40 ]
Мы представляем экспериментальные результаты для этой реакции в виде двух химических шагов:
| S 1 + EH → P 1 + EP 2 | ( 3 ) |
| EP 2 + H -O -H → EH + P 2 | ( 4 ) |
Где S 1 является полипептидом, P 1 и P 2 являются продуктами. Первая химическая стадия ( 3 ) включает в себя образование ковалентного ацил-анзимента. Второй шаг ( 4 ) - это шаг деахилирования. Важно отметить, что группа H+, первоначально обнаруженная на ферменте, но не в воде, появляется в продукте до стадии гидролиза, поэтому она может рассматриваться как дополнительная группа ферментативной реакции.
Таким образом, реакция ( 3 ) показывает, что фермент действует как мощный реагент реакции. Согласно предлагаемой концепции, H -транспорт из фермента способствует первой конверсии реагента, разбив первой первоначальной химической связи (между группами P 1 и P 2 ). Стадия гидролиза приводит к разрушению второй химической связи и регенерации фермента.
Предлагаемый химический механизм не зависит от концентрации субстратов или продуктов в среде. Однако сдвиг в их концентрации в основном вызывает изменения свободной энергии на первой и последней стадии реакций ( 1 ) и ( 2 ) из -за изменений содержания свободной энергии в каждой молекуле, будь то S или P, в водном растворе. Этот подход соответствует следующему механизму сокращения мышц. Последним этапом гидролиза АТФ в скелетных мышцах является высвобождение продукта, вызванное ассоциацией головок миозина с актином. [ 41 ] Закрытие актин-связывающей расщелины во время реакции ассоциации структурно связано с открытием нуклеотид-связывающего кармана на активном месте миозина. [ 42 ]
Примечательно, что последние этапы гидролиза АТФ включают быстрое высвобождение фосфата и медленное высвобождение ADP. [ 43 ] [ 44 ] Высвобождение фосфатного аниона из связанного аниона ADP в водный раствор может рассматриваться как эксергоническая реакция, поскольку фосфатный анион имеет низкую молекулярную массу.
Таким образом, мы приходим к выводу, что первичное высвобождение неорганического фосфата H 2 PO 4 − Приводит к трансформации значительной части свободной энергии гидролиза АТФ в кинетическую энергию сольватированного фосфата, создавая активную потоковую передачу. Это предположение о локальной механической химической трансдукции соответствует механизму сокращения мышц Тироша, где мышечная сила вытекает из интегрированного действия активного потокового вещания, созданного гидролизом АТФ. [ 45 ] [ 46 ]
Примеры каталитических механизмов
[ редактировать ]В действительности, большинство ферментных механизмов включают в себя комбинацию нескольких различных типов катализа.
Триозофосфат изомераза
[ редактировать ]Триозофосфат изомераза EC 5.3.1.1 ) катализирует обратимую взаимосвязь двух триозофосфатов ( изомеров дигидроксиацетонового фосфата и d- глицеральдегида 3-фосфата .
Трипсин
[ редактировать ]Трипсин ( EC 3.4.21.4 ) представляет собой сериновую протеазу , которая расщепляет белковые субстраты после лизина или аргинина остатков с использованием каталитической триады для выполнения ковалентного катализа, и отверстие оксианиона для стабилизации заряда в переходных состояниях .
Альдолаза
[ редактировать ]Альдолаза ( EC 4.1.2.13 ) катализирует распад фруктозы 1,6-бисфосфата (F-1,6-пт) в глицеральдегид 3-фосфат и дигидроксиацетон фосфат ( DHAP ).
Фермент диффузию
[ редактировать ]Появление одномолекулярных исследований в 2010-х годах привело к наблюдению, что движение неподвижных ферментов увеличивается с увеличением концентрации субстрата и увеличивая энтальпию реакции . [ 47 ] Последующие наблюдения показывают, что это увеличение диффузии обусловлено переходным смещением центра масс фермента , что приводит к «эффекту отдачи, который вызывает фермент». [ 48 ]
Сходство реакции
[ редактировать ]Сходство между ферментативными реакциями ( EC ) может быть рассчитано с использованием изменений связей, реакционных центров или метрик субструктуры ( AC-Blast Archived 30 мая 2019 года на машине Wayback ). [ 49 ]
Смотрите также
[ редактировать ]- Каталитическая триада
- Анализ ферментов
- Ингибитор фермента
- Кинетика фермента
- Распущенность фермента
- Протеиновая динамика
- Псевдоэнзименты , чье вездесущность, несмотря на их каталитическую бездействие, предполагает омические последствия
- Квантовое туннелирование
- Карта протеолиза
- Время разрешило кристаллографию
Ссылки
[ редактировать ]- ^ Kamerlin SC, Warshel A (май 2010 г.). «На рассвете 21 -го века: является ли динамика отсутствующим звеном для понимания ферментного катализа?» Полем Белки . 78 (6): 1339–1375. doi : 10.1002/prot.22654 . PMC 2841229 . PMID 20099310 .
- ^ Лейдлер К.Дж. (1978). Физическая химия с биологическим применением . Бенджамин/Каммингс. п. 427. ISBN 978-0-8053-5680-9 .
- ^ Кошленд де (февраль 1958 г.). «Применение теории ферментной специфичности к синтезу белка» . Труды Национальной академии наук Соединенных Штатов Америки . 44 (2): 98–104. Bibcode : 1958pnas ... 44 ... 98K . doi : 10.1073/pnas.44.2.98 . PMC 335371 . PMID 16590179 .

- ^ Анслин Э.В., Догерти Д.А. (2006). Современная физическая органическая химия . Университетские научные книги. ISBN 978-1-891389-31-3 .
- ^ Савир Y, Tlusty T (май 2007 г.). Scalas E (ред.). «Конформационная корректура: влияние конформационных изменений на специфичность молекулярного распознавания» . Plos один . 2 (5): E468. Bibcode : 2007ploso ... 2..468s . doi : 10.1371/journal.pone.0000468 . PMC 1868595 . PMID 17520027 .
- ^ Stanton RV, Peräkylä M, Bakowies D, Kollman PA (1998). «Комбинированные расчеты AB initio и свободной энергии для изучения реакций в ферментах и растворе: амидный гидролиз в трипсине и водном растворе». J. Am. Химический Соц 120 (14): 3448–3457. doi : 10.1021/ja972723x .
- ^ Кун Б., Коллман П.А. (2000). «Расчеты QM-FE и молекулярной динамики на катехол о-метилтрансфераза: свободная энергия активации в ферменте и в водном растворе и региоселективность реакции, катализируемой ферментами». J. Am. Химический Соц 122 (11): 2586–2596. doi : 10.1021/ja992218v .
- ^ Bruice TC, Lightstone FC (1999). «Основное состояние и вклад в переходное состояние в показатели внутримолекулярных и ферментативных реакций». Акк. Химический Резерв 32 (2): 127–136. doi : 10.1021/ar960131y .
- ^ Page MI, Jencks WP (август 1971 г.). «Энтропийные вклад в ускорения скорости в ферментативных и внутримолекулярных реакциях и эффект хелата» . Труды Национальной академии наук Соединенных Штатов Америки . 68 (8): 1678–1683. Bibcode : 1971pnas ... 68.1678p . doi : 10.1073/pnas.68.8.1678 . PMC 389269 . PMID 5288752 .
- ^ Warshel A, Parson WW (ноябрь 2001 г.). «Динамика биохимических и биофизических реакций: понимание компьютерного моделирования». Ежеквартальные обзоры биофизики . 34 (4): 563–679. doi : 10.1017/s0033583501003730 . PMID 11852595 . S2CID 28961992 .
- ^ Jump up to: а беременный в дюймовый Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MH (август 2006 г.). «Электростатическая основа для ферментного катализа». Химические обзоры . 106 (8): 3210–3235. doi : 10.1021/cr0503106 . PMID 16895325 .
- ^ Warshel A, Naray-Szabo G, Sussman F, Hwang JK (май 1989). «Как действительно работают сериновые протеазы?». Биохимия . 28 (9): 3629–3637. doi : 10.1021/bi00435a001 . PMID 2665806 .
- ^ Fersht AR, Requena Y (декабрь 1971 г.). «Механизм, катализируемый -химотрипсин -гидролиз амидов. Зависимость от рН от KC и K M. Кинетическое обнаружение промежуточного соединения». Журнал Американского химического общества . 93 (25): 7079–7087. doi : 10.1021/ja00754a066 . PMID 5133099 .
- ^ Zeeberg B, Caswell M, Caplow M (апрель 1973 г.). «Относительно сообщаемого изменения стадии определения скорости в катализе химотрипсина». Журнал Американского химического общества . 95 (8): 2734–2735. doi : 10.1021/ja00789a081 . PMID 4694533 .
- ^ Foot D, Foot JG (2011). Биохимия . Джон Уайли и сыновья. OCLC 808679090 .
- ^ Маркус Р.А. (1965). «О теории реакций переноса электронов. VI. Единое лечение гомогенных и электродных реакций» (PDF) . J. Chem. Физический 43 (2): 679–701. Bibcode : 1965jchph..43..679m . doi : 10.1063/1.1696792 .
- ^ Warshel A (ноябрь 1978 г.). «Энергетика ферментного катализа» . Труды Национальной академии наук Соединенных Штатов Америки . 75 (11): 5250–5254. Bibcode : 1978pnas ... 75.5250W . doi : 10.1073/pnas.75.11.5250 . PMC 392938 . PMID 281676 .
- ^ Fried SD, Bagchi S, Boxer SG (декабрь 2014 г.). «Экстремальные электрические поля мощности катализ в активном участке кетостероидной изомеразы» . Наука . 346 (6216). Нью -Йорк, Нью -Йорк: 1510–4. BIBCODE : 2014SCI ... 346.1510F . doi : 10.1126/science.1259802 . PMC 4668018 . PMID 25525245 .
- ^ Toney, MD «специфичность реакции в пиридоксальных ферментах». Архивы биохимии и биофизики (2005) 433: 279-287
- ^ «Информационный центр микроэлементов, Университет штата Орегон» . Архивировано из оригинала 21 марта 2015 года . Получено 30 сентября 2009 года .
- ^ Voet D, Voet JG (2004). Биохимия John Wiley & Sounds Inc. стр. 986–989 . ISBN 978-0-471-25090-6 .
- ^ Voet D, Voet JG (2004). Биохимия John Wiley & Sounds Inc. стр. 604–606 . ISBN 978-0-471-25090-6 .
- ^ Piccirilli JA, Vyle JS, Caruthers MH, Cech TR (январь 1993 г.). «Катализ ионов металлов в реакции рибозима тетрагимена». Природа . 361 (6407): 85–88. Bibcode : 1993natur.361 ... 85p . doi : 10.1038/361085A0 . PMID 8421499 . S2CID 4326584 .
- ^ Бендер М.Л. (1 января 1962 г.). «Катализ ионов металлов нуклеофильных органических реакций в растворе». Реакции скоординированных лигандов . Достижения в области химии. Тол. 37. Американское химическое общество. С. 19–36. doi : 10.1021/ba-1963-0037.ch002 . ISBN 978-0-8412-0038-8 .
- ^ Fife TH, Przystas TJ (1 февраля 1985 г.). «Дивалентный ионный катализ металла в гидролизе сложных эфиров пиколиновой кислоты. Ион металла способствовал ионам гидроксида и катализируемых водяными реакциями». Журнал Американского химического общества . 107 (4): 1041–1047. doi : 10.1021/ja00290a048 . ISSN 0002-7863 .
- ^ Stadtman ER (1 января 1990 г.). «Катализируемое металлом окисление белков: биохимический механизм и биологические последствия» . Свободная радикальная биология и медицина . 9 (4): 315–325. doi : 10.1016/0891-5849 (90) 90006-5 . PMID 2283087 .
- ^ Jencks WP (1987) [1969]. Катализ в химии и фермере . Серия McGraw-Hill в Advanced Chemistry (Перепечатка изд.). Нью -Йорк: Dover Publications . ISBN 978-0-486-65460-7 .
- ^ Warshel A, Levitt M (май 1976 г.). «Теоретические исследования ферментативных реакций: диэлектрическая, электростатическая и стерическая стабилизация иона карбона в реакции лизоцима». Журнал молекулярной биологии . 103 (2): 227–249. doi : 10.1016/0022-2836 (76) 90311-9 . PMID 985660 .
- ^ Voet D, Voet JG, Pratt CW (2013). Основы биохимии: жизнь на молекулярном уровне (четвертое изд.). Хобокен, Нью -Джерси: Уайли. ISBN 978-0-470-54784-7 .
- ^ Garcia-Viloca M, Gao J, Karplus M, Truhlar DG (январь 2004 г.). «Как работают ферменты: анализ по современной теории скорости и компьютерному моделированию». Наука . 303 (5655): 186–195. Bibcode : 2004sci ... 303..186G . doi : 10.1126/science.1088172 . PMID 14716003 . S2CID 17498715 .
- ^ Jump up to: а беременный Олссон М.Х., Зигбан П.Е., Варшель А (март 2004 г.). «Моделирование большого кинетического изотопного эффекта и температурной зависимости переноса атома водорода в липоксигеназе». Журнал Американского химического общества . 126 (9): 2820–2828. doi : 10.1021/ja037233l . PMID 14995199 .
- ^ Jump up to: а беременный Масграу Л., Руджеиникова А., Йоханниссен Л.О., Хоти П., Басран Дж, Ранаган К.Е. и др. (Апрель 2006 г.). «Атомное описание ферментной реакции, в которой преобладает протонное туннелирование». Наука . 312 (5771): 237–241. Bibcode : 2006sci ... 312..237M . doi : 10.1126/science.1126002 . PMID 16614214 . S2CID 27201250 .
- ^ Hwang JK, Warshel A (1996). «Насколько важны квантовые механические ядерные движения в ферментативном катализе». J. Am. Химический Соц 118 (47): 11745–11751. doi : 10.1021/ja962007f .
- ^ Ball P (сентябрь 2004 г.). "Ферменты: случайно или по дизайну?" Полем Природа . 431 (7007): 396–397. Bibcode : 2004natur.431..396b . doi : 10.1038/431396a . PMID 15385982 . S2CID 228263 .
- ^ Olsson MH, Parson WW, Warshel A (май 2006 г.). «Динамический вклад в ферментный катализ: критические тесты популярной гипотезы». Химические обзоры . 106 (5): 1737–1756. doi : 10.1021/cr040427e . PMID 16683752 .
- ^ Vol'kenshtein MV, Dogonadze RR, Madumarov AK, Urushadze ZD, Kharkats Yi (1972). «Теория ферментного катализа». Молекулярная биология . 6 (3). Москва: 347–353. PMID 4645409 .
- ^ Volkenshtein MV, Dogonadze RR, Madumarov AK, Urushadze ZD, Kharkats Yu I (1973). "Electronic and Conformational Interactions in Enzyme Catalysis.". Konformatsionnie Izmenenia Biopolimerov v Rastvorakh . Moscow: Nauka Publishing House. pp. 153–157.
- ^ Foigel AG (июнь 2011 г.). «Является ли фермент мощным реагентом биохимической реакции?». Молекулярная и клеточная биохимия . 352 (1–2): 87–89. doi : 10.1007/s11010-011-0742-4 . PMID 21318350 . S2CID 11133081 .
- ^ Fogel AG (август 1982). «Кооперативность ферментативных реакций и молекулярные аспекты энергетической трансдукции». Молекулярная и клеточная биохимия . 47 (1): 59–64. doi : 10.1007/bf00241567 . PMID 7132966 . S2CID 21790380 .
- ^ Hengge AC, Stein RL (январь 2004 г.). «Роль конформационной мобильности белка в ферментном катализе: ацилирование альфа-химотрипсина специфическими пептидными субстратами». Биохимия . 43 (3): 742–747. doi : 10.1021/bi030222k . PMID 14730979 .
- ^ Lymn RW, Taylor EW (декабрь 1971 г.). «Механизм аденозин -трифосфатного гидролиза актомиозином». Биохимия . 10 (25): 4617–4624. doi : 10.1021/bi00801a004 . PMID 4258719 .
- ^ Холмс К.С., Ангит I, Кулл Ф.Дж., Ян В., Шредер Р.Р. (сентябрь 2003 г.). «Электронная крио-микроскопия показывает, как сильное связывание миозина с актином высвобождает нуклеотид». Природа . 425 (6956): 423–427. Bibcode : 2003natur.425..423H . doi : 10.1038/nature02005 . PMID 14508495 . S2CID 2686184 .
- ^ Siemankowski RF, Wiseman MO, White HD (февраль 1985 г.). «Диссоциация ADP от подтефрагмента актомиозина 1 достаточно медленная, чтобы ограничить выгруженную скорость укорочения в мышцах позвоночных» . Труды Национальной академии наук Соединенных Штатов Америки . 82 (3): 658–662. Bibcode : 1985pnas ... 82..658s . doi : 10.1073/pnas.82.3.658 . PMC 397104 . PMID 3871943 .
- ^ White HD, Belknap B, Webb MR (сентябрь 1997 г.). «Кинетика нуклеозидного трифосфатного расщепления и этапов высвобождения фосфата с помощью ассоциированного скелетного актемиозина кролика, измеренного с использованием нового флуоресцентного зонда для фосфата». Биохимия . 36 (39): 11828–11836. doi : 10.1021/bi970540h . PMID 9305974 .
- ^ Тирош Р., Лоу Вз, Оплатка А (март 1990 г.). «Трансляционное движение актиновых филаментов в присутствии тяжелого меромиозина и MGATP, измеренных с помощью допплеровского расширения лазерного рассеяния света». Biochimica et Biophysica Acta (BBA) - структура белка и молекулярная ферма . 1037 (3): 274–280. doi : 10.1016/0167-4838 (90) 90025-b . PMID 2178685 .
- ^ Тирош Р. (2006). «Баллистические протоны и микроволновые растворы воды (солитоны) в биоэнергетических преобразовании» . Инт. J. Mol. Наука 7 (9): 320–345. doi : 10.3390/i7090320 .
- ^ Muddana HS, Sengupta S, Mallouk TE, Sen A, Butler PJ (февраль 2010 г.). «Субстратный катализ усиливает диффузию одномерных» . Журнал Американского химического общества . 132 (7): 2110–2111. doi : 10.1021/ja908773a . PMC 2832858 . PMID 20108965 .

- ^ Ридель С., Габизон Р., Уилсон К.А., Хамадани К., Цекурас К., Маркузи С. и др. (Январь 2015). «Тепло, выделяемое во время каталитического оборота, усиливает диффузию фермента» . Природа . 517 (7533): 227–230. Bibcode : 2015natur.517..227r . doi : 10.1038/nature14043 . PMC 4363105 . PMID 25487146 .
- ^ Rahman SA, Cuesta SM, Furnham N, Holliday GL, Thornton JM (февраль 2014 г.). «EC-Blast: инструмент для автоматического поиска и сравнения реакций ферментов» . Природные методы . 11 (2): 171–174. doi : 10.1038/nmeth.2803 . PMC 4122987 . PMID 24412978 .
Дальнейшее чтение
[ редактировать ]- Fersht A (1998). Структура и механизм в белковой науке: руководство по катализу фермента и складывания белка . Нью -Йорк: WH Freeman. ISBN 978-0-7167-3268-6 .
- Сатклифф М., Мунро А (август 2006 г.). «Квантовой катализ в ферментах - с теорией переходного состояния» . Философские транзакции б . 361 (1472): 1291–1455. doi : 10.1098/rstb.2006.1879 . PMC 1647302 .
Внешние ссылки
[ редактировать ] СМИ, связанные с ферментным катализом в Wikimedia Commons
СМИ, связанные с ферментным катализом в Wikimedia Commons
| Базы данных управления авторитетом : национальный |
|---|