Трансмиссивная губчатая энцефалопатия
Эта статья требует внимания эксперта в области медицины . Конкретная проблема заключается в следующем: раздел «Причина» находится в довольно плохом состоянии и имеет чрезмерный вес. ( январь 2022 г. ) |
| Трансмиссивная губчатая энцефалопатия (TSE) | |
|---|---|
| Другие имена | Прионовая болезнь |
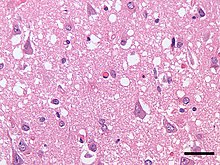 | |
| Микрофотография, показывающая губчатую дегенерацию ( вакуоли , которые выглядят как отверстия в срезах ткани) в коре головного мозга пациента, умершего от болезни Крейтцфельдта-Якоба . Окраска H&E , масштабная линейка = 30 микрон (0,03 мм). | |
| Специальность | Инфекционные заболевания |
| Симптомы | Деменция, судороги, тремор, бессонница, психоз, делирий, спутанность сознания. |
| Обычное начало | От месяцев до десятилетий |
| Типы | Губчатая энцефалопатия крупного рогатого скота , Фатальная семейная бессонница , болезнь Крейцфельдта-Якоба , куру , болезнь Хантингтона 1 , скрепи , вариабельно-протеазо-чувствительная прионопатия , хроническая истощающая болезнь , синдром Герстмана-Штраусслера-Шейнкера , губчатая энцефалопатия кошек , трансмиссивная энцефалопатия норки , экзотические копытные энцефалопатия , верблюжья губчатая энцефалопатия |
| Причины | Прион |
| Факторы риска | Контакт с инфицированными жидкостями, проглатывание зараженной плоти, наличие заболевания у одного или двух родителей (в случае фатальной семейной бессонницы) |
| Метод диагностики | В настоящее время не существует другого способа надежно обнаружить прионы, кроме как при вскрытии. |
| Профилактика | Варьируется |
| Уход | Паллиативная помощь |
| Прогноз | Неизменно фатальный |
| Частота | Редкий |
Трансмиссивные губчатые энцефалопатии ( ТГЭ ), также известные как прионные болезни , [1] представляют собой группу прогрессирующих, неизлечимых и смертельных заболеваний, связанных с прионами и поражающих мозг и нервную систему многих животных , включая человека , крупный рогатый скот и овец . По наиболее распространенной гипотезе, они передаются прионами , хотя некоторые другие данные позволяют предположить причастность спироплазменной инфекции. [2] Умственные и физические способности ухудшаются, и в коре головного мозга появляется множество крошечных отверстий, из-за чего она выглядит как губка, когда ткань мозга, полученная при аутопсии, исследуется под микроскопом . Расстройства вызывают нарушение функции мозга, что может привести к потере памяти, изменениям личности, а также ненормальным или нарушенным движениям , которые со временем ухудшаются. [3]
ТГЭ у людей включают болезнь Крейтцфельдта-Якоба , синдром Герстмана-Штраусслера-Шейнкера , фатальную семейную бессонницу и куру , а также недавно обнаруженную прионопатию с различной протеазной чувствительностью и семейную губчатую энцефалопатию. Сама болезнь Крейтцфельдта-Якоба имеет четыре основные формы: спорадическую (сБКЯ), наследственную/семейную (фБКЯ), ятрогенную (иБКЯ) и вариантную форму (вБКЯ). Эти состояния образуют спектр заболеваний с перекрывающимися признаками и симптомами.
TSE у млекопитающих, кроме человека, включает скрепи у овец, губчатую энцефалопатию крупного рогатого скота (BSE) у крупного рогатого скота, широко известную как «коровье бешенство», и хроническую истощающую болезнь (CWD) у оленей и лосей. Вариантная форма болезни Крейтцфельдта-Якоба у человека вызвана воздействием прионов губчатой энцефалопатии крупного рогатого скота . [4] [5] [6]
В отличие от других видов инфекционных заболеваний, которые распространяются агентами с геномом ДНК или РНК (например, вирусами или бактериями ), инфекционный агент ТГЭ считается прионом , то есть состоит исключительно из белкового материала. Неправильно свернутые прионные белки переносят заболевание между людьми и вызывают ухудшение состояния мозга . ТГЭ являются уникальными заболеваниями, поскольку их этиология может быть генетической, спорадической или инфекционной при употреблении в пищу инфицированных пищевых продуктов или ятрогенными способами (например, переливание крови). [7] Большинство ТГЭ являются спорадическими и возникают у животных без мутаций прионного белка. Наследственный ТГЭ встречается у животных, несущих редкий мутантный приона аллель , вызывающую заболевание , который экспрессирует прионные белки, которые сами по себе искажаются в конформацию . Передача происходит, когда здоровые животные потребляют зараженные ткани других больных. В 1980-е и 1990-е годы губчатая энцефалопатия крупного рогатого скота приобрела эпидемический характер. Это произошло потому, что крупный рогатый скот кормили переработанными остатками другого крупного рогатого скота , и эта практика сейчас запрещена во многих странах. В свою очередь, потребление (человеком) пищевых продуктов бычьего происхождения, содержащих ткани, загрязненные прионами, привело к вспышке вариантной формы болезни Крейтцфельдта-Якоба в 1990-х и 2000-х годах. [8]
Прионы не могут передаваться по воздуху, через прикосновения или при большинстве других форм случайных контактов. Однако они могут передаваться при контакте с инфицированными тканями, жидкостями организма или загрязненными медицинскими инструментами. Обычные процедуры стерилизации , такие как кипячение или облучение материалов, не могут сделать прионы неинфекционными. Однако обработка сильным, почти неразбавленным отбеливателем и/или гидроксидом натрия или нагревание до температуры минимум 134 °C действительно разрушает прионы. [9]
Классификация
[ редактировать ]Различия в форме между различными формами прионного белка плохо изучены.
| Код ICTVdb [10] | Название болезни | Естественный хозяин | Название приона | PrP Изоформа | Жвачное животное |
|---|---|---|---|---|---|
| Нечеловеческие млекопитающие | |||||
| 90.001.0.01.001. | Скрэпи | Овцы и козы | Скрепи прион | ПрП наук | Да |
| 90.001.0.01.002. | Трансмиссивная норковая энцефалопатия (ТМЕ) | Норка | ТМЕ прион | ПрП ТМЕ | Нет |
| 90.001.0.01.003. | Хроническая истощающая болезнь (ХИЗ) | Лось , белохвостый олень , олень-мул и благородный олень | CWD прион | ПрП УВП | Да |
| 90.001.0.01.004. | Губчатая энцефалопатия крупного рогатого скота (ГЭКРС) широко известное как «коровье бешенство» | Крупный рогатый скот | прион ГЭКРС | ПрП БФБ | Да |
| 90.001.0.01.005. | Губчатая энцефалопатия кошек (FSE) | Кошки | ФСЕ прион | ПрП ФСЕ | Нет |
| 90.001.0.01.006. | Экзотическая энцефалопатия копытных (ЭЭЭ) | Ньяла и больший куду | ЭУЭ прион | ПрП Евросоюз | Да |
| Верблюжья губчатая энцефалопатия (CSE) [11] | Верблюд | ПрП СSE | Да | ||
| Болезни человека | |||||
| 90.001.0.01.007. | Кому | Люди | Какой прион | ПрП Кому | Нет |
| 90.001.0.01.008. | Болезнь Крейтцфельдта-Якоба (БКЯ) | прионы БКЯ | ПрП СКЖД | Нет | |
| Вариант болезни Крейтцфельдта-Якоба (вБКЯ, нвБКЯ) | vCJD прион [12] | ПрП vCJD | |||
| 90.001.0.01.009. | Синдром Герстмана-Штраусслера-Шейнкера (СГШ) | ГСС прион | ПрП ГСС | Нет | |
| 90.001.0.01.010. | Фатальная семейная бессонница (FFI) | FFI прион | ПрП ИФИ | Нет | |
| Семейная губчатая энцефалопатия [13] | |||||
Признаки и симптомы
[ редактировать ]Дегенеративное поражение тканей, вызванное прионными заболеваниями человека (БКЯ, ГСС и куру), характеризуется четырьмя признаками: губкообразным изменением (наличием множества мелких отверстий), гибелью нейронов , астроцитозом (аномальным увеличением числа астроцитов вследствие разрушение близлежащих нейронов) и образование амилоидных бляшек. Эти черты являются общими с прионными заболеваниями у животных, и признание этих сходств побудило к первым попыткам передать приону человека прионную болезнь (куру) приматам в 1966 году, за которыми последовали CJD в 1968 году и GSS в 1981 году. Эти нейропатологические особенности сформировались. легли в основу гистологической диагностики прионных заболеваний человека на протяжении многих лет, хотя было признано, что эти изменения чрезвычайно вариабельны как от случая к случаю, так и внутри центральной нервной системы в отдельных случаях. [14]
Клинические признаки у людей различаются, но обычно включают изменения личности, психиатрические проблемы, такие как депрессия , нарушение координации и/или неустойчивая походка ( атаксия ). Пациенты также могут испытывать непроизвольные подергивания, называемые миоклонусами , необычные ощущения, бессонницу , спутанность сознания или проблемы с памятью. На поздних стадиях заболевания у пациентов наблюдаются тяжелые психические нарушения ( деменция ) и они теряют способность двигаться и говорить. [15]
Ранние невропатологические сообщения о прионных заболеваниях человека страдали от путаницы в номенклатуре, в которой иногда упускалось из виду значение диагностического признака губчатых изменений. Последующая демонстрация того, что прионные заболевания человека являются трансмиссивными, подтвердила важность губчатых изменений как диагностического признака, что отражено в использовании термина «губчатая энцефалопатия» для этой группы заболеваний.
Прионы наиболее заразны при прямом контакте с пораженными тканями. Например, болезнь Крейтцфельдта-Якоба передавалась пациентам, получавшим инъекции гормона роста человека , полученного из гипофиза , из аллотрансплантатов твердой мозговой оболочки трупа и из инструментов, используемых в операциях на головном мозге (Brown, 2000) (прионы могут пережить в « автоклаве используемый процесс стерилизации »). для большинства хирургических инструментов). Также считается [ кем? ] что употребление в пищу больных животных может привести к медленному накоплению прионов, особенно когда каннибализм или аналогичные практики позволяют белкам накапливаться в течение более чем одного поколения. Примером является куру , которая достигла масштабов эпидемии в середине 20-го века у народа Форе в Папуа-Новой Гвинее , который употреблял в пищу своих мертвецов в качестве погребального ритуала. [16] В настоящее время законы в развитых странах запрещают использование переработанных белков жвачных животных в кормах для жвачных животных в качестве меры предосторожности против распространения прионной инфекции среди крупного рогатого скота и других жвачных животных. [ нужна ссылка ]
Имеются данные о том, что прионные заболевания могут передаваться воздушно-капельным путем. [17]
Обратите внимание, что не все энцефалопатии вызваны прионами, как в случае ПМЛ (вызванного вирусом JC ), CADASIL (вызванного аномальной активностью белка NOTCH3) и болезни Краббе (вызванной дефицитом фермента галактозилцерамидазы ). Прогрессирующая губчатая лейкоэнцефалопатия (ПСЛ), которая представляет собой губчатую энцефалопатию, также, вероятно, не вызывается прионом, хотя прион, вызывающий ее у курильщиков героина, еще не идентифицирован. [18] [19] [20] [21] Это, в сочетании с весьма разнообразной природой патологии прионных заболеваний, является причиной того, что прионную болезнь нельзя диагностировать только на основании симптомов пациента.
Причина
[ редактировать ]Генетика
[ редактировать ]Мутации PRNP гена вызывают прионную болезнь. Семейные формы прионных заболеваний вызваны наследственными мутациями гена PRNP. Однако лишь небольшой процент всех случаев прионового заболевания передается в семьях. Большинство случаев прионных заболеваний являются спорадическими, то есть возникают у людей без каких-либо известных факторов риска или генных мутаций. В редких случаях прионные заболевания также могут передаваться при контакте с тканями, загрязненными прионами, или другими биологическими материалами, полученными от людей с прионными заболеваниями.
Ген PRNP дает инструкции по созданию белка, называемого прионным белком (PrP). В нормальных условиях этот белок может участвовать в транспортировке меди в клетки. Белок также может участвовать в защите клеток мозга и помогать им общаться. [22] [23] Точечные мутации в этом гене заставляют клетки вырабатывать аномальную форму прионного белка, известную как PrP. наук . Этот аномальный белок накапливается в мозге и разрушает нервные клетки, что приводит к появлению признаков и симптомов прионной болезни.
Семейные формы прионных заболеваний наследуются по аутосомно-доминантному типу, что означает, что одной копии измененного гена в каждой клетке достаточно, чтобы вызвать заболевание. В большинстве случаев больной человек наследует измененный ген от одного больного родителя.
У некоторых людей семейные формы прионной болезни вызваны новой мутацией гена PRNP. Хотя у таких людей, скорее всего, нет пострадавшего родителя, они могут передать генетическое изменение своим детям.
Белковая гипотеза
[ редактировать ]Белок может быть инфекционным агентом, индуцирующим собственную репликацию, вызывая конформационные изменения нормального клеточного PrP. С в ПрП наук . Доказательства этой гипотезы:
- Титр инфекционности коррелирует с PrP наук уровни. Однако это оспаривается. [24]
- ПрП наук является изомером PrP С
- Денатурация PrP устраняет инфекционность [25]
- Мыши с нулевым PrP не могут быть инфицированы [26]
- ПрП С истощение нервной системы мышей с установленной нейроинвазивной прионной инфекцией обращает вспять ранний спонгиоз и поведенческие нарушения, останавливает дальнейшее прогрессирование заболевания и увеличивает продолжительность жизни [27]
Многокомпонентная гипотеза
[ редактировать ]Хотя прионы не содержат генома нуклеиновой кислоты, они могут состоять не только из белка. Очищенный ПрП С кажется, неспособен преобразоваться в инфекционный PrP наук форме, если не добавлены другие компоненты, такие как РНК и липиды. [28] Эти другие компоненты, называемые кофакторами, могут составлять часть инфекционного приона или служить катализаторами репликации белкового приона.
Вирусная гипотеза
[ редактировать ]Эта гипотеза постулирует, что причиной заболевания является еще не обнаруженный инфекционный вирусный агент. Хотя когда-то эта гипотеза была основной, сейчас ее придерживается меньшинство. Доказательства этой гипотезы следующие:
- Время инкубации сравнимо с лентивирусом .
- Вариации штаммов различных изолятов PrP СК . [29]
Диагностика
[ редактировать ]По-прежнему существует очень практическая проблема с диагностикой прионных заболеваний, включая BSE и CJD. У них есть инкубационный период от месяцев до десятилетий, в течение которого симптомы отсутствуют, хотя путь превращения нормального белка PrP мозга в токсичный, связанный с заболеванием PrP наук форма началась. В настоящее время практически не существует способов обнаружения PrP. наук достоверно, кроме как при исследовании головного мозга нейропатологическими и иммуногистохимическими методами после смерти. Накопление аномально свернутого PrP наук Форма белка PrP является характеристикой заболевания, но она присутствует в очень низких количествах в легкодоступных жидкостях организма, таких как кровь или моча. Исследователи попытались разработать методы измерения PrP. наук , но до сих пор не существует полностью признанных методов использования таких материалов, как кровь. [ нужна ссылка ]
В 2010 году команда из Нью-Йорка описала обнаружение PrP. наук даже если изначально он присутствует в количестве лишь одной стомиллиардной (10 −11 ) в ткани головного мозга. Этот метод сочетает в себе амплификацию с новой технологией под названием Surround Optical Fiber Immunoassay (SOFIA) и некоторыми специфическими антителами против PrP. наук . После амплификации и последующей концентрации любого PrP наук , образцы метят флуоресцентным красителем с использованием антитела для специфичности, а затем, наконец, загружают в микрокапиллярную трубку. Эта трубка помещается в специально сконструированное устройство так, что она полностью окружена оптическими волокнами для улавливания всего света, излучаемого при возбуждении красителя с помощью лазера. Методика позволила обнаружить PrP. наук после гораздо меньшего количества циклов конверсии, чем было достигнуто другими, что существенно снижает вероятность появления артефактов, а также ускоряет анализ. Исследователи также проверили свой метод на образцах крови внешне здоровых овец, у которых впоследствии развилась скрепи. Мозг животных анализировали, как только проявлялись какие-либо симптомы. Таким образом, исследователи смогли сравнить результаты тканей головного мозга и крови, взятых после того, как у животных проявились симптомы заболеваний, с кровью, полученной ранее в жизни животных, и от неинфицированных животных. Результаты очень ясно показали, что PrP наук можно было обнаружить в крови животных задолго до появления симптомов. [30] [31]
Уход
[ редактировать ]В настоящее время не известны способы лечения или предотвращения прионных заболеваний. [32] Некоторые лекарства могут замедлить прогрессирование заболевания. Но в конечном итоге поддерживающая терапия — единственный вариант для людей на данный момент.
Эпидемиология
[ редактировать ]Трансмиссивная губчатая энцефалопатия (ТГЭ) встречается очень редко, но может достигать масштабов эпидемии. [ нужны разъяснения ] Очень сложно составить карту распространения заболевания из-за сложности идентификации отдельных штаммов прионов. Это означает, что если у животных на одной ферме заболевание начинает проявляться после вспышки на соседней ферме, очень трудно определить, является ли это один и тот же штамм, поражающий оба стада (что предполагает передачу), или же вторая вспышка произошла из-за совершенно другой источник. [ нужна ссылка ]
Классическая болезнь Крейтцфельдта-Якоба (БКЯ) была открыта в 1920 году. Она встречается спорадически во всем мире, но очень редко. Ежегодно от него страдает примерно один человек на миллион. Обычно в этих случаях причина неизвестна. Было обнаружено, что в некоторых случаях он передается генетически. 250 пациентов заразились заболеванием ятрогенным путем ( при использовании зараженного хирургического оборудования). [33] Это было до того, как в 1976 году потребовалась стерилизация оборудования, и с тех пор других ятрогенных случаев не было. Чтобы предотвратить распространение инфекции, Всемирная организация здравоохранения создала руководство, в котором рассказывает медицинским работникам, что делать при появлении БКЯ и как утилизировать зараженное оборудование. [34] Центры по контролю и профилактике заболеваний (CDC) ведут наблюдение за случаями CJD, в частности, просматривая информацию в свидетельствах о смерти. [35]
Хроническая истощающая болезнь (ХИБ) — это прионное заболевание, обнаруженное в Северной Америке у оленей и лосей. Первый случай был идентифицирован как синдром смертельного истощения в 1960-х годах. Затем в 1978 году это заболевание было признано трансмиссивной губчатой энцефалопатией. Исследования показали, что эндемическое заболевание CWD у оленей и лосей, находящихся на свободном выгуле, распространено на северо-востоке Колорадо, юго-востоке Вайоминга и западной Небраске. Также было обнаружено, что CWD мог присутствовать у части животных, находящихся на свободном выгуле, за десятилетия до первоначального признания. В Соединенных Штатах открытие CWD вызвало обеспокоенность по поводу передачи этого прионного заболевания человеку. Во многих очевидных случаях CJD подозревалась передача CWD, однако доказательства отсутствовали и не были убедительными. [36]
В 1980-х и 1990-х годах губчатая энцефалопатия крупного рогатого скота (ГЭКРС или «коровье бешенство») распространялась среди крупного рогатого скота с эпидемической скоростью. Общее предполагаемое количество инфицированного крупного рогатого скота в период с 1980 по 1996 год составило около 750 000. Это произошло потому, что крупный рогатый скот кормили обработанными остатками другого крупного рогатого скота. Затем потребление человеком этого зараженного скота вызвало вспышку человеческой формы CJD. После введения запрета на кормление произошло резкое снижение заболеваемости ГЭКРС. 20 мая 2003 г. первый случай ГЭКРС был подтвержден в Северной Америке. Источник не удалось точно определить, но исследователи подозревают, что он произошел из импортированного коровьего мяса, инфицированного ГЭКРС. В Соединенных Штатах Министерство сельского хозяйства США создало меры безопасности, чтобы свести к минимуму риск заражения людей ГЭКРС. [37]
Вариант болезни Крейтцфельдта-Якоба (вБКЯ) был обнаружен в 1996 году в Англии. Имеются убедительные доказательства того, что вБКЯ вызвана тем же прионом, что и губчатая энцефалопатия крупного рогатого скота. [38] Всего с момента его первого обнаружения был зарегистрирован 231 случай vCJD. Эти случаи были обнаружены в общей сложности в 12 странах: 178 в Великобритании, 27 во Франции, пять в Испании, четыре в Ирландии, четыре в США, три в Нидерландах, три в Италии, два в Португалии, два в Канаде и по одному в Японии, Саудовской Аравии и Тайване. [39]
История
[ редактировать ]В V веке эры до нашей Гиппократ описал такое заболевание, как ТГЭ, у крупного рогатого скота и овец, которое, по его мнению, также встречается у людей. [40] Публий Флавий Вегетий Ренат записывает случаи заболевания со схожими характеристиками в IV и V веках нашей эры. [41] В 1755 году вспышка скрепи обсуждалась в Британской палате общин и, возможно, существовала в Британии некоторое время до этого. [42] Хотя в 1759 году существовали необоснованные утверждения о том, что болезнь заразна, в целом считалось, что она возникла из-за инбридинга, и контрмеры оказались успешными. Эксперименты начала 20-го века не смогли доказать передачу скрепи между животными, пока не были приняты чрезвычайные меры, такие как внутриглазная инъекция инфицированной нервной ткани. Никакой прямой связи между скрепи и заболеванием человека тогда не подозревалось и с тех пор не было обнаружено. TSE был впервые описан у людей Альфонсом Марией Якобом в 1921 году. [43] Открытие Дэниела Карлтона Гайдусека о том, что Куру передался каннибализмом, сопровождающееся обнаружением скрепи-подобных поражений в мозгу жертв Куру, убедительно свидетельствует об инфекционной основе ТГЭ. [44] Сдвиг парадигмы в сторону ненуклеиновой инфекционной сущности потребовался, когда результаты были подтверждены объяснением того, как прионный белок может передавать губчатую энцефалопатию. [45] Лишь в 1988 году невропатология губчатой энцефалопатии была должным образом описана у коров. [46] Тревожное распространение ГЭ в британском стаде крупного рогатого скота усилило страх перед передачей вируса человеку и укрепило веру в инфекционную природу ТГЭ. Это было подтверждено выявлением , болезни Куру, названной новым вариантом болезни Крейтцфельдта-Якоба у людей, подвергшихся воздействию ГЭКРС . [47] Хотя модель инфекционного заболевания ТГЭ была подвергнута сомнению в пользу модели прионной трансплантации, которая объясняет, почему каннибализм способствует передаче инфекции, [48] По состоянию на 2007 год поиск вирусного агента продолжался в некоторых лабораториях. [49] [50]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ «Трансмиссивные губчатые энцефалопатии» . Национальный институт неврологических расстройств и инсульта . Проверено 23 апреля 2023 г.
- ^ Бастиан Ф.О. , Сандерс Д.Э., Форбс В.А., Хагиус С.Д., Уокер Дж.В., Хенк В.Г., Энрайт Ф.М., Эльзер П.Х. (2007). « Spiroplasma spp. из трансмиссивной губчатой энцефалопатии головного мозга или клещей вызывает губчатую энцефалопатию у жвачных животных» . Журнал медицинской микробиологии . 56 (9): 1235–1242. дои : 10.1099/jmm.0.47159-0 . ПМИД 17761489 .
- ^ Чесебро, Брюс (1 июня 2003 г.). «Введение в трансмиссивные губчатые энцефалопатии или прионные заболевания». Британский медицинский бюллетень . 66 (1): 1–20. дои : 10.1093/bmb/66.1.1 . ISSN 1471-8391 . ПМИД 14522845 .
- ^ «Вариант болезни Крейцфельдта-Якоба» . Всемирная организация здравоохранения . Проверено 25 апреля 2017 г. Февраль 2012 г. Архивировано из оригинала 20 декабря 2002 г.
- ^ «Вариант болезни Крейцфельдта-Якоба > Связь с ГЭКРС (коровье бешенство)» . Центры по контролю и профилактике заболеваний . Проверено 25 апреля 2017 г. 10 февраля 2015 г.
- ^ Коллиндж, Дж; Сидле, КЦ; Мидс, Дж; Айронсайд, Дж; Хилл, AF (24 октября 1996 г.). «Молекулярный анализ вариаций прионного штамма и этиологии« нового варианта »БКЯ». Природа . 383 (6602): 685–690. Бибкод : 1996Natur.383..685C . дои : 10.1038/383685a0 . ПМИД 8878476 . S2CID 4355186 .
- ^ Браун П., Прис М., Брандел Дж.П., Сато Т., МакШейн Л., Зерр И., Флетчер А., Уилл Р.Г., Покьяри М., Кэшман Н.Р., д'Эно Дж.Х., Червенакова Л., Фрадкин Дж., Шонбергер Л.Б., Коллинз С.Дж. (2000). «Ятрогенная болезнь Крейцфельдта-Якоба в тысячелетии». Неврология . 55 (8): 1075–81. дои : 10.1212/WNL.55.8.1075 . ПМИД 11071481 . S2CID 25292433 .
- ^ Колле, Дж.Г.; Брэдли, Р; Либерский, ПП (2006). «Вариант CJD (vCJD) и губчатая энцефалопатия крупного рогатого скота (BSE): 10 и 20 лет спустя: часть 2». Фолиа Нейропатолика . 44 (2): 102–110. ПМИД 16823692 .
- ^ «Варианты прионной дезинфекции – биобезопасность и гигиена труда» . Университет Миннесоты . 17 ноября 2017 г.
- ^ «ICTVdB: универсальная вирусная база данных Международного комитета по таксономии вирусов — Каталог NLM — NCBI» . www.ncbi.nlm.nih.gov . Проверено 23 апреля 2023 г.
- ^ «Два алжирских исследователя обнаружили в Уаргле «бешеную верблюжью болезнь»» . 09.05.2018. Архивировано из оригинала 17 июня 2018 г. Проверено 13 марта 2019 г.
- ^ Считается, что он идентичен приону BSE.
- ^ Нитрини Р., Роземберг С., Пассос-Буэно М.Р., да Силва Л.С., Югетти П., Пападопулос М., Каррильо П.М., Карамелли П., Альбрехт С., Затц М., ЛеБлан А. (август 1997 г.). «Семейная губчатая энцефалопатия, связанная с новой мутацией гена прионного белка». Анналы неврологии 42 (2): 138–46. дои : 10.1002/ana.410420203 . ПМИД 9266722 . S2CID 22600579 .
- ^ Джеффри М., Гудбранд И.А., Гудсир К.М. (1995). «Патология трансмиссивных губчатых энцефалопатий с особым акцентом на ультраструктуру». Микрон . 26 (3): 277–98. дои : 10.1016/0968-4328(95)00004-N . ПМИД 7788281 .
- ^ Коллиндж Дж. (2001). «Прионные болезни человека и животных: их причины и молекулярные основы». Анну преподобный Neurosci . 24 : 519–50. дои : 10.1146/annurev.neuro.24.1.519 . ПМИД 11283320 .
- ^ Коллинз С., Маклин Калифорния, Мастерс CL (2001). «Синдром Герстмана-Штрауслера-Шейнкера, фатальная семейная бессонница и куру: обзор этих менее распространенных трансмиссивных губчатых энцефалопатий человека». Дж. Клин Неврология . 8 (5): 387–97. дои : 10.1054/jocn.2001.0919 . ПМИД 11535002 . S2CID 31976428 .
- ^ Хайбек, Йоханнес; Хейкенвальдер, Матиас; Клевенц, Бритта; Блэк, Петра; Маргалит, Илан; Бридель, Клэр; Мерц, Кирстен; Зирдум, Элизабет; Петч, Бенджамин; Фукс, Томас Дж.; Ститц, Лотар; Агуцци, Адриано (13 января 2011 г.). «Аэрозоли передают прионы иммунокомпетентным и иммунодефицитным мышам» . ПЛОС Патогены . 7 (1): e1001257. дои : 10.1371/journal.ppat.1001257 . ПМК 3020930 . ПМИД 21249178 .
- ^ «hafci.org» . Архивировано из оригинала 1 ноября 2004 года . Проверено 2 декабря 2007 г.
- ^ Кригштейн А.Р.; Шунгу, округ Колумбия; Миллар В.С.; и др. (1999). «Лейкоэнцефалопатия и повышение уровня лактата в мозге от вдыхания паров героина («погоня за драконом»)». Неврология . 53 (8): 1765–73. дои : 10.1212/WNL.53.8.1765 . ПМИД 10563626 . S2CID 2915734 .
- ^ Чанг YJ, Цай CH, Чен CJ (1997). «Лейкоэнцефалопатия после вдыхания паров героина». Дж. Формос. Мед. доц . 96 (9): 758–60. ПМИД 9308333 .
- ^ Кусса С., Забад Р., Ризк Т., Тамраз Дж., Наснас Р., Чемали Р. (2002). «[Героин-индуцированная вакуолярная лейкоэнцефалопатия: 4 случая]». Преподобный о. Нейрол. (Париж) (на французском языке). 158 (2): 177–82. ПМИД 11965173 .
- ^ Сакудо, Акиказу; Ли, Дыг-чан; Саэки, Кейичи; Накамура, Юко; Иноуэ, Кейичи; Мацумото, Ёсицугу; Итохара, Сигэёси; Онодера, Такаши (2003). «Нарушение активации супероксиддисмутазы усеченным на N-конце прионным белком (PrP) в линии нейрональных клеток с дефицитом PrP». Связь с биохимическими и биофизическими исследованиями . 308 (3): 660–667. дои : 10.1016/s0006-291x(03)01459-1 . ISSN 0006-291X . ПМИД 12914801 .
- ^ Коллиндж, Джон; Уиттингтон, Майлз А.; Сидл, Кэти CL; Смит, Корин Дж.; Палмер, Марк С.; Кларк, Энтони Р.; Джефферис, Джон Г. Р. (1994). «Прионный белок необходим для нормальной синаптической функции» . Природа . 370 (6487): 295–297. Бибкод : 1994Natur.370..295C . дои : 10.1038/370295a0 . ISSN 1476-4687 . ПМИД 8035877 .
- ^ Бэррон Р.М.; Кэмпбелл С.Л.; Кинг Д; и др. (декабрь 2007 г.). «Высокие титры инфекционной трансмиссивной губчатой энцефалопатии, связанные с чрезвычайно низкими уровнями PrPSc in vivo» . Журнал биологической химии . 282 (49): 35878–86. дои : 10.1074/jbc.M704329200 . ПМИД 17923484 .
- ^ Супаттапон С; Вилле Х; Уечи Л; и др. (апрель 2001 г.). «Разветвленные полиамины лечат прион-инфицированные клетки нейробластомы» . Журнал вирусологии . 75 (7): 3453–61. doi : 10.1128/JVI.75.7.3453-3461.2001 . ПМЦ 114138 . ПМИД 11238871 .
- ^ Сакудо А; Ли, округ Колумбия; Саэки К; и др. (август 2003 г.). «Нарушение активации супероксиддисмутазы усеченным на N-конце прионным белком (PrP) в линии нейрональных клеток с дефицитом PrP». Связь с биохимическими и биофизическими исследованиями . 308 (3): 660–7. дои : 10.1016/S0006-291X(03)01459-1 . ПМИД 12914801 .
- ^ Маллуччи Дж; Дикинсон А; Линехэм Дж; и др. (октябрь 2003 г.). «Истощение нейронального PrP при прионной инфекции предотвращает заболевание и обращает вспять спонгиоз». Наука . 302 (5646): 871–874. Бибкод : 2003Sci...302..871M . дои : 10.1126/science.1090187 . ПМИД 14593181 . S2CID 13366031 .
- ^ Делео Н.Р., Харрис Б.Т., Рис-младший, Супаттапоне С. (июнь 2007 г.). «Формирование нативных прионов из минимальных компонентов in vitro» . Труды Национальной академии наук Соединенных Штатов Америки . 104 (23): 9741–6. Бибкод : 2007PNAS..104.9741D . дои : 10.1073/pnas.0702662104 . ПМК 1887554 . ПМИД 17535913 .
- ^ Брюс М.Э. (2003). «Вариация штамма TSE» . Британский медицинский бюллетень . 66 : 99–108. дои : 10.1093/bmb/66.1.99 . ПМИД 14522852 .
- ^ «Обнаружение прионов в крови» (PDF) . Микробиология сегодня. : 195. Август 2010 г. Архивировано из оригинала (PDF) 31 марта 2012 г. Проверено 21 августа 2011 г.
- ^ «SOFIA: Аналитическая платформа для сверхчувствительного обнаружения PrP наук в мозге и крови» (PDF) . Медицинский центр SUNY Downstate . Проверено 19 августа 2011 г. .
- ^ «Терапевтические подходы к прионовым заболеваниям | NIAID: Национальный институт аллергии и инфекционных заболеваний» . www.niaid.nih.gov . 21 октября 2019 г. Проверено 17 июля 2024 г.
- ^ «Трансмиссивные губчатые энцефалопатии (ТГЭ), также известные как прионные заболевания | Anses - Национальное агентство по безопасности пищевых продуктов, окружающей среды и гигиены труда» . www.anses.fr . 18 февраля 2013 года . Проверено 9 ноября 2017 г.
- ^ «Инфекционный контроль | Классическая болезнь Крейтцфельдта-Якоба (CJD) | Прионная болезнь | CDC» . www.cdc.gov . Проверено 9 ноября 2017 г.
- ^ «Наблюдение за vCJD | Классический вариант болезни Крейтцфельдта-Якоба (CJD) | Прионная болезнь | CDC» . www.cdc.gov . Проверено 9 ноября 2017 г.
- ^ Белай и Шенбергер (2005). «Влияние прионных заболеваний на общественное здравоохранение» (PDF) . Ежегодный обзор общественного здравоохранения . 26 : 206–207. doi : 10.1146/annurev.publhealth.26.021304.144536 . ПМИД 15760286 .
- ^ Белай и Шенбергер (2005). «Влияние прионных заболеваний на общественное здравоохранение» (PDF) . Ежегодный обзор общественного здравоохранения . 26 : 198–201. doi : 10.1146/annurev.publhealth.26.021304.144536 . ПМИД 15760286 .
- ^ «Вариант болезни Крейцфельдта-Якоба» . Всемирная организация здравоохранения. Архивировано из оригинала 20 декабря 2002 года . Проверено 9 ноября 2017 г.
- ^ «Риск для путешественников | Вариант болезни Крейцфельдта-Якоба, классический (БКЯ) | Прионная болезнь» . www.cdc.gov . Проверено 9 ноября 2017 г.
- ^ Макалистер, В. (июнь 2005 г.). «Священная болезнь нашего времени: провал инфекционной модели губчатой энцефалопатии» . Клин Инвест Мед . 28 (3): 101–4. ПМИД 16021982 . Проверено 20 июня 2011 г.
- ^ Дигеста искусства медицины мулов , Публий Флавий Вегетий Ренат
- ^ Браун П., Брэдли Р.; Брэдли (декабрь 1998 г.). «1755 год и все такое: исторический учебник трансмиссивной губчатой энцефалопатии» . БМЖ . 317 (7174): 1688–92. дои : 10.1136/bmj.317.7174.1688 . ПМЦ 1114482 . ПМИД 9857129 .
- ^ Качер Ф. (май 1998 г.). «Это болезнь Якоба, а не Крейцфельдта» . Природа . 393 (6680): 11. Бибкод : 1998Natur.393Q..11K . дои : 10.1038/29862 . ПМИД 9590681 . S2CID 205000018 .
- ^ Гайдусек, округ Колумбия (сентябрь 1977 г.). «Нетрадиционные вирусы, происхождение и исчезновение куру». Наука . 197 (4307): 943–60. Бибкод : 1977Sci...197..943C . дои : 10.1126/science.142303 . ПМИД 142303 .
- ^ Коллинз С.Дж., Лоусон В.А., магистр CL.; Лоусон; Мастера (январь 2004 г.). «Трансмиссивные губчатые энцефалопатии». Ланцет . 363 (9204): 51–61. дои : 10.1016/S0140-6736(03)15171-9 . ПМИД 14723996 . S2CID 23212525 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Хоуп Дж., Рики Л.Дж., Хантер Н., Мультауп Г., Бейройтер К., Уайт Х., Скотт А.С., Стек М.Дж., Доусон М., Уэллс Г.А.; Рики; Хантер; Мультауп; Бейройтер; Белый; Скотт; Куча; Доусон; и др. (ноябрь 1988 г.). «Фибриллы из мозга коров с новым заболеванием крупного рогатого скота содержат белок, связанный со скрепи». Природа . 336 (6197): 390–2. Бибкод : 1988Natur.336..390H . дои : 10.1038/336390a0 . ПМИД 2904126 . S2CID 4351199 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Уилл Р.Г., Айронсайд Дж.В., Зейдлер М., Казенс С.Н., Эстибейро К., Альперович А., Позер С., Поккиари М., Хофман А., Смит П.Г.; Айронсайд; Зейдлер; Казенс; Эстибейро; Альперович; Позер; Поккьяри; Хофман; Смит (апрель 1996 г.). «Новый вариант болезни Крейтцфельдта-Якоба в Великобритании». Ланцет . 347 (9006): 921–5. дои : 10.1016/S0140-6736(96)91412-9 . ПМИД 8598754 . S2CID 14230097 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Макалистер, В. (июнь 2005 г.). «Священная болезнь нашего времени: провал инфекционной модели губчатой энцефалопатии» . Клин Инвест Мед . 28 (3): 101–4. ПМИД 16021982 . Проверено 20 июня 2011 г.
- ^ Мануэлидис Л , Ю ЗХ, Баркеро Н, Банкеро Н, Маллинз Б; Ю; Банкеро; Маллинз (февраль 2007 г.). «Клетки, инфицированные скрепи и возбудителями болезни Крейцфельдта-Якоба, производят внутриклеточные вирусоподобные частицы размером 25 нм» . Труды Национальной академии наук Соединенных Штатов Америки . 104 (6): 1965–70. Бибкод : 2007ПНАС..104.1965М . дои : 10.1073/pnas.0610999104 . ПМК 1794316 . ПМИД 17267596 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ «Инфекционные частицы» . Лаборатория Мануэлидиса .
- Эта запись включает в себя общедоступный текст, первоначально предоставленный Национальным институтом неврологических расстройств и инсульта, Национальными институтами здравоохранения [1] и Национальной медицинской библиотекой США [2].
Внешние ссылки
[ редактировать ]| Базы данных органов управления : Национальные |
|---|