Болезнь Урбаха-Вите
| Болезнь Урбаха-Вите | |
|---|---|
| Другие имена | Липоидный протеиноз и гиалиноз кожи и слизистых оболочек. |
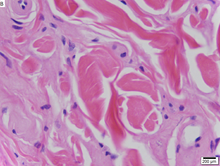 | |
| Болезнь Урбаха-Вите при биопсии кожи с окраской H&E . | |
| Специальность | Эндокринология |

Болезнь Урбаха-Вите — очень редкое рецессивное генетическое заболевание, с момента его открытия зарегистрировано около 400 случаев. [ 1 ] [ 2 ] [ 3 ] Впервые об этом официально сообщили в 1929 году Эрих Урбах и Камилло Вите . [ 4 ] [ 5 ] хотя можно признать случаи, датированные еще 1908 годом. [ 6 ] [ 7 ] [ 8 ]
Симптомы заболевания сильно варьируются от человека к человеку. Они могут включать хриплый голос, поражения и рубцы на коже, легко повреждаемую кожу с плохим заживлением ран, сухую, морщинистую кожу и образование папул вокруг век. [ 6 ] [ 9 ] [ 10 ] Все это является результатом общего утолщения кожи и слизистых оболочек . В ряде случаев наблюдается также уплотнение мозговой ткани в медиальных височных долях , что может привести к эпилепсии и нервно-психическим отклонениям. [ 11 ] Заболевание обычно не опасно для жизни, и у пациентов не наблюдается снижения продолжительности жизни . [ 10 ]
Поскольку болезнь Урбаха-Вите является аутосомно-рецессивным заболеванием, люди могут быть носителями заболевания, но не проявлять никаких симптомов. потери функции Заболевание вызвано мутациями хромосомы 1 в 1q21, гена белка внеклеточного матрикса 1 ( ECM1 ). [ 12 ] Дерматологические симптомы вызваны накоплением гиалинового материала в дерме и утолщением базальных мембран кожи. [ 9 ] Болезнь Урбаха-Вите обычно диагностируется по клиническим дерматологическим проявлениям, особенно по папулам в виде бусинок на веках. Открытие мутаций в гене ECM1 позволило использовать генетическое тестирование для подтверждения первоначального клинического диагноза . периодическую кислоту-Шиффа (PAS) и иммуногистохимическое окрашивание. Для диагностики также можно использовать [ 6 ] [ 13 ]
В настоящее время не существует лекарства от болезни Урбаха-Вите, хотя существуют способы индивидуального лечения многих ее симптомов. [ 2 ] Открытие мутаций гена ECM1 открыло возможность генной терапии или использования рекомбинантного белка ECM1 для лечения болезни Урбаха-Вите, но ни один из этих вариантов в настоящее время недоступен. Некоторые исследователи обследуют пациентов с болезнью Урбаха-Вите, чтобы узнать больше о других состояниях, которые проявляют сходные неврологические симптомы, таких как аутизм .
Симптомы и признаки
[ редактировать ]Болезнь Урбаха-Вите характеризуется как неврологической , так и дерматологической симптоматикой. [ 14 ] [ 15 ]
Дерматологический
[ редактировать ]Хотя симптомы могут сильно различаться у разных людей, даже у членов одной семьи, симптомы обычно начинаются в младенчестве и обычно являются результатом утолщения кожи и слизистых оболочек. [ 2 ] Первым симптомом часто является слабый крик или осипший голос из-за утолщения голосовых связок. Охриплость голоса может быть одним из наиболее ярких клинических проявлений заболевания. [ 9 ] Поражения и шрамы также появляются на коже , обычно на лице и дистальных частях конечностей. [ 6 ] Часто это является результатом плохого заживления ран, и по мере старения пациента рубцы продолжают увеличиваться, придавая коже восковой вид. Кожу можно легко повредить в результате незначительной травмы или травмы, оставляя множество волдырей и дополнительных шрамов. [ 10 ] Кожа также обычно очень сухая и морщинистая. образуются белые или желтые инфильтраты . На губах, слизистой оболочке щек , миндалинах , язычке , надгортаннике и уздечке языка [ 6 ] Это может привести к инфекции верхних дыхательных путей , и иногда требуется трахеостомия . для облегчения симптомов [ 9 ] Слишком сильное утолщение уздечки может ограничить движение языка и привести к нарушениям речи . [ 16 ] Образование папул вокруг век распространенным симптомом и часто используется как часть диагностики заболевания является очень . Некоторые другие дерматологические симптомы, которые иногда наблюдаются, но менее распространены, включают выпадение волос , паротит и другие стоматологические аномалии, изъязвление роговицы и очаговую дегенерацию макулы . [ 17 ]
Неврологический
[ редактировать ]Хотя дерматологические изменения являются наиболее очевидными симптомами болезни Урбаха-Вите, у многих пациентов также наблюдаются неврологические симптомы. Около 50–75% диагностированных случаев болезни Урбаха-Вите также обнаруживают двусторонние симметричные кальцинаты в медиальных височных долях . [ 18 ] Эти кальцификации часто поражают миндалевидное тело и периамигдалоидные извилины . [ 11 ] Считается, что миндалевидное тело участвует в обработке биологически значимых стимулов и эмоциональной долговременной памяти, особенно тех, которые связаны со страхом , а ПЭТ и МРТ показали корреляцию между активацией миндалевидного тела и эпизодической памятью для сильно эмоциональных стимулов. [ 14 ] Следовательно, пациенты с болезнью Урбаха-Вите с кальцификациями и поражениями в этих областях могут страдать от нарушений в этих системах. Эти кальцификации являются результатом накопления отложений кальция в кровеносных сосудах этой области мозга. Со временем эти сосуды твердеют , и ткань, частью которой они являются, отмирает, вызывая повреждения. Количество кальцификации часто связано с продолжительностью заболевания. [ 10 ] Истинную распространенность этих кальцификатов трудно точно определить, поскольку не всем пациентам проводится визуализация головного мозга . У некоторых пациентов также наблюдаются эпилепсия и нервно-психические нарушения. Симптомы эпилепсии могут начинаться с легких приступов тревоги, и их можно контролировать с помощью противоэпилептических препаратов. [ 10 ] У других пациентов наблюдаются симптомы, похожие на шизофрению, а некоторые страдают от расстройств настроения, тревоги и психотических расстройств. [ 16 ] [ 19 ]

Причины
[ редактировать ]Исследователи сопоставили болезнь Урбаха-Вите с хромосомой 1 на участке 1q21 и конкретно идентифицировали ген белка 1 внеклеточного матрикса ( ECM1 ) как ген, содержащий мутации, которые могут привести к развитию этого заболевания. [ 12 ] На данный момент сообщалось, что 41 различная мутация в ECM1 приводит к болезни Урбаха-Вите. [ 13 ] Все это были гомозиготные мутации с потерей функции (т.е. нонсенс , сдвиг рамки считывания или внутренние делеции ). [ 9 ] Это аутосомно-рецессивное заболевание, [ 2 ] [ 13 ] для возникновения заболевания требуются две мутированные копии гена ECM1. [ 20 ]
ECM1 кодирует гликопротеин ранее неизвестного происхождения. Открытие того, что потеря экспрессии ECM1 приводит к симптомам, связанным с болезнью Урбаха-Вите, предполагает, что ECM1 может способствовать адгезии кожи, эпидермальной дифференцировке, а также заживлению ран и образованию рубцов. [ 12 ] Также считается, что он играет роль в формировании эндохондральной кости, биологии опухолей, пролиферации эндотелиальных клеток и формировании кровеносных сосудов. [ 9 ]
Дерматологические симптомы вызваны накоплением гиалинового материала в дерме и утолщением базальных мембран кожи. [ 9 ] Природа этого материала неизвестна, но исследователи предположили, что это может быть гликопротеин, гликолипид , кислый мукополисахарид , измененный коллаген или эластическая ткань. [ 6 ]
Диагностика
[ редактировать ]Болезнь Урбаха-Вите обычно диагностируется по клиническим дерматологическим проявлениям, особенно по папулам в виде бусинок на веках. Врачи также могут проверить гиалиновый материал с помощью периодического окрашивания кислотой Шиффа (PAS), поскольку при этом окрашивании материал сильно окрашивается. [ 6 ]
Было показано, что иммуногистохимическое маркирование кожи антителами к белку ECM1 в качестве маркировки снижается в коже людей, пораженных болезнью Урбаха-Вите. [ 13 ] Окрашивание антителами к коллагену типа IV или антителами к коллагену типа VII выявляет яркие толстые полосы на дермоэпидермальном соединении . [ 9 ]
без контрастирования КТ позволяет визуализировать кальцификаты, но обычно не используется в качестве средства диагностики заболевания. Частично это связано с тем, что не у всех пациентов Урбаха-Вите наблюдаются кальцификаты, но также и с тем, что подобные поражения могут образовываться при других заболеваниях, таких как простой герпес и энцефалит . Открытие мутаций в гене ECM1 позволило использовать генетическое тестирование для подтверждения первоначального клинического диагноза болезни Урбаха-Вите. Это также позволяет врачам лучше отличать болезнь Урбаха-Вите от других подобных заболеваний, не вызванных мутациями ECM1. [ нужна ссылка ]
Уход
[ редактировать ]В настоящее время не существует лекарства от болезни Урбаха-Вите, хотя существуют способы индивидуального лечения многих ее симптомов. Некоторый успех был достигнут при пероральном применении диметилсульфоксида (ДМСО) и внутриочаговом гепарине , но это верно не во всех случаях. [ 9 ] [ 19 ] D- пеницилламин также показал многообещающие результаты, но пока не получил широкого применения. [ 13 ] Есть также некоторые сообщения о пациентах, которых лечили этретинатом , препаратом, который обычно назначают для лечения псориаза . [ 17 ] В некоторых случаях кальцификаты в головном мозге могут привести к аномальной электрической активности нейронов. Некоторым пациентам назначают противосудорожные препараты, чтобы помочь справиться с этими отклонениями. Трахеостомию часто используют для облегчения инфекций верхних дыхательных путей. Углекислотная лазерная хирургия утолщенных голосовых связок и папул век с бусинами улучшила эти симптомы у пациентов. [ 9 ] Открытие мутаций гена ECM1 открыло возможность генной терапии или использования рекомбинантного белка EMC1 для лечения болезни Урбаха-Вите, но ни один из этих двух вариантов в настоящее время недоступен. [ нужна ссылка ]
Прогноз
[ редактировать ]Болезнь Урбаха-Вите обычно не является опасным для жизни состоянием. [ 2 ] Ожидаемая продолжительность жизни этих пациентов является нормальной, если должным образом устраняются потенциальные побочные эффекты утолщения слизистой оболочки, такие как обструкция дыхательных путей. [ 10 ] Хотя для этого может потребоваться трахеостомия или лазерная операция на углекислом газе, такие шаги могут помочь гарантировать, что люди с болезнью Урбаха-Вите смогут жить полноценной жизнью. Было показано, что пероральный диметилсульфоксид (ДМСО) уменьшает повреждения кожи, помогая минимизировать дискомфорт для этих людей. [ 9 ]
Заболеваемость
[ редактировать ]Болезнь Урбаха-Вите встречается очень редко; описано менее 300 случаев в медицинской литературе . [ 2 ] Хотя болезнь Урбаха-Вите можно обнаружить во всем мире, почти четверть зарегистрированных диагнозов приходится на Южную Африку . [ 2 ] Многие из них наблюдаются у пациентов голландского , немецкого и койсанского происхождения. [ 2 ] [ 12 ] Считается, что такая высокая частота обусловлена эффектом основателя . [ 13 ] Из-за рецессивной генетической причины и способности быть переносчиком заболевания без симптомов болезнь Урбаха-Вите часто передается в семьях. В некоторых регионах Южной Африки переносчиком заболевания может быть до одного из 12 человек. [ 9 ] Большинство тематических исследований с участием пациентов с болезнью Урбаха-Вите включают только один-три случая, и эти случаи часто происходят в одной семье. Из-за низкой заболеваемости трудно найти достаточно большое количество случаев для адекватного изучения заболевания. [ нужна ссылка ]
История
[ редактировать ]В 1908 году о первом случае болезни Урбаха-Вите сообщил Фридрих Зибенманн , профессор отоларингологии из Базеля , Швейцария . В 1925 году Фридрих Мишер , швейцарский дерматолог, сообщил о трех подобных пациентах. [ 6 ] Официальное сообщение о болезни Урбаха-Вите было впервые описано в 1929 году венским дерматологом и оториноларингологом Урбахом и Вите. [ 12 ] Его первоначальное название «липоидоз кожи и слизистых оболочек» было изменено на «липоидный протеиноз кожи и слизистых оболочек» из-за убеждения Урбаха, что это состояние возникает из-за аномальных отложений липидов и белков в тканях. [ 12 ] Некоторые спорят о том, является ли это заболевание на самом деле формой мукополисахаридоза , амилоидоза или даже порфирии . Открытие болезни Урбаха-Вите, вызывающей мутацию гена ECM1, теперь предоставило окончательный способ дифференцировать болезнь Урбаха-Вите от этих других состояний. [ 9 ]
См. также
[ редактировать ]- Синдром Клювера-Бьюси
- СМ (пациент)
- Список кожных заболеваний
- Список рентгенографических данных, связанных с кожными заболеваниями
Ссылки
[ редактировать ]- ^ «Познакомьтесь с женщиной, которая не чувствует страха — The Washington Post» . Вашингтон Пост .
- ^ Jump up to: а б с д и ж г час ДиДжандоменико С.; Маси Р.; Кассандрини Д.; Эль-Хашем М.; ДеВито Р.; Бруно К.; Санторелли FM (2006). «Липоидный протеиноз: описание случая и обзор литературы» . Акта Оториноларингол Итал . 26 (3): 162–7. ПМК 2639960 . ПМИД 17063986 .
- ^ Джеймс, Уильям Д.; Бергер, Тимоти Г.; и др. (2006). Болезни кожи Эндрюса: клиническая дерматология . Сондерс Эльзевир. ISBN 978-0-7216-2921-6 .
- ^ Synd/924 в Who Named It?
- ^ Урбах Э., Вите К. (1929). «Липоидоз кожи и слизистых оболочек» . Архив Вирхова по патологической анатомии и физиологии и клинической медицине . 273 (2): 285–319. дои : 10.1007/bf02158983 . S2CID 42016927 .
- ^ Jump up to: а б с д и ж г час Каро I (1978). «Липоидный протеиноз». Международный журнал дерматологии . 17 (5): 388–93. дои : 10.1111/ijd.1978.17.5.388 . ПМИД 77850 . S2CID 43544386 .
- ^ Левер, Уолтер Ф.; Старейшина, Дэвид А. (2005). Гистопатология кожи по Леверу . Хагерствон, доктор медицины: Липпинкотт Уильямс и Уилкинс . п. 440. ИСБН 978-0-7817-3742-5 .
- ^ Зибенманн Ф. (1908). «О вовлечении слизистой оболочки в общий гиперкератоз кожи». Арка Ларингол . 20 :101-109.
- ^ Jump up to: а б с д и ж г час я дж к л м Хамада, Т. (2002). «Липоидный протеиноз». Клиническая и экспериментальная дерматология . 27 (8): 624–629. дои : 10.1046/j.1365-2230.2002.01143.x . ПМИД 12472532 . S2CID 28344373 .
- ^ Jump up to: а б с д и ж Аппенцеллер, С; Шалу, Э; Велью, П; Де Соуза, ЕМ; Араужо, ВЗ; Сендес, Ф; Ли, Л.М. (2006). «Кальцификация миндалины, связанная с продолжительностью заболевания при липоидном протеинозе». Журнал нейровизуализации . 16 (2): 154–156. дои : 10.1111/j.1552-6569.2006.00018.x . ПМИД 16629738 . S2CID 30567332 .
- ^ Jump up to: а б Стаут, CCV; Найдич, Т.П. (1998). «Болезнь Урбаха-Вите (липоидный протеиноз)». Детская нейрохирургия . 28 (4): 212–214. дои : 10.1159/000028653 . ПМИД 9732251 . S2CID 46862405 .
- ^ Jump up to: а б с д и ж Хамада Т.; Маклин WHI; Рамзи М.; Эштон GHS; Нанда А.; и др. (2002). «Липоидный протеиноз картируется 1q21 и вызван мутациями в гене белка 1 внеклеточного матрикса (ECM1)». Молекулярная генетика человека . 11 (7): 833–40. дои : 10.1093/hmg/11.7.833 . ПМИД 11929856 .
- ^ Jump up to: а б с д и ж Чан И.; Лю Л.; Хамада Т.; Сетураман Г.; МакГрат Дж. А. (2007). «Молекулярная основа липоидного протеиноза: мутации в белке 1 внеклеточного матрикса» . Экспериментальная дерматология . 16 (11): 881–90. дои : 10.1111/j.1600-0625.2007.00608.x . ПМИД 17927570 .
- ^ Jump up to: а б Зиберт М.; Маркович Х.Дж.; Бартель П. (2003). «Миндалина, аффект и познание: данные 10 пациентов с болезнью Урбаха-Вите» . Мозг . 126 (12): 2627–37. дои : 10.1093/brain/awg271 . ПМИД 12937075 .
- ^ Мэллори С.Б.; Крафчик Б.Р.; Холм С.А.; Ленейн П.; Крафчик Б.Р. (2005). «Что это за синдром? Синдром Урбаха-Вейте (липоидный протеиноз)». Детская дерматология . 22 (3): 266–7. дои : 10.1111/j.1525-1470.2005.22321.x . ПМИД 15916581 . S2CID 44925736 .
- ^ Jump up to: а б Торнтон HB; Нел Д.; Торнтон Д.; ван Хонк Дж.; Бейкер Дж.А.; Штейн DJ (2008). «Нейропсихиатрия и нейропсихология липоидного протеиноза» . Журнал нейропсихиатрии и клинических нейронаук . 20 (1): 86–92. дои : 10.1176/jnp.2008.20.1.86 . ПМИД 18305289 .
- ^ Jump up to: а б Бахадир С.; Чобаньоглу У.; Капичиоглу З.; Свеча СТ; Чимсит Г.; и др. (2006). «Липоидный протеиноз: случай с офтальмологическими и психиатрическими данными». Журнал дерматологии 33 (3): 215–8. дои : 10.1111/j.1346-8138.2006.00049.x . ПМИД 16620230 . S2CID 34699559 .
- ^ Хурлеманн Р.; Вагнер М.; Хавеллек Б.; Райх Х.; Пиперхофф П.; Амунц К.; и др. (2007). «Контроль миндалевидного тела забыванием и запоминанием, вызванным эмоциями: данные болезни Урбаха-Вите». Нейропсихология . 45 (5): 877–84. doi : 10.1016/j.neuropsychologia.2006.08.027 . hdl : 21.11116/0000-0001-B8EF-3 . ПМИД 17027866 . S2CID 4101263 .
- ^ Jump up to: а б Чиназ П.; Гувенир Т.; Гонлусен Г. (1993). «Липоидный протеиноз: болезнь Урбаха-Вите». Акта Педиатрика . 82 (11): 892–3. дои : 10.1111/j.1651-2227.1993.tb12590.x . ПМИД 8241657 . S2CID 31104438 .
- ^ Мороввати С., Фаршадьегане П., Хамидизаде М., Мороввати З., Мохаммади С.Д. (август 2018 г.). «Анализ мутаций гена ECM1 у двух родственных иранских пациентов, страдающих липоидным протеинозом» . Акта Медика Ираника . 56 (7): 474–477 . Проверено 1 сентября 2020 г.