Несовершенный остеогенез
| Несовершенный остеогенез (НО) | |
|---|---|
| Другие имена | болезнь хрупких костей, [1] синдром Лобштейна, [1] : 5 хрупкость костей [2] веселая болезнь, [1] : 5 идиопатический остеопсатироз [3] : 347 |
 | |
| Синие склеры являются классическим непатогномоничным признаком несовершенного остеогенеза. | |
| Произношение | |
| Специальность | Педиатрия , медицинская генетика , ортопедия |
| Симптомы | Кости легко ломаются , синий оттенок склер ( белков глаз), небольшой рост, гипермобильность суставов , потеря слуха. [5] |
| Начало | Рождение |
| Продолжительность | Долгосрочная перспектива |
| Причины | Генетическая ( аутосомно-доминантная мутация или de novo мутация ) [6] |
| Метод диагностики | На основании симптомов проводится ДНК-тестирование. |
| Профилактика | Преимплантационная генетическая диагностика |
| Управление | Здоровый образ жизни (занятия спортом, отказ от курения), металлические стержни через длинные кости. |
| Медикамент | Бисфосфонаты [7] |
| Прогноз | Зависит от типа |
| Частота | 1 из 15 000–20 000 человек [8] |


Несовершенный остеогенез (англ. IPA : / ˌ ɒ s t i oʊ ˈ dʒ ɛ n ə s ɪ s ˌ ɪ m p ɜːr ˈ f ɛ k t ə / ; [4] OI ), в просторечии известный как болезнь хрупких костей , представляет собой группу генетических нарушений , которые приводят к тому кости , что легко ломаются . [1] : 85 [9] Диапазон симптомов – как на скелете тела , так и на других органах – может быть от легкой до тяжелой. [5] : 1512 Симптомы, обнаруживаемые при различных типах ОИ, включают белки глаз (склеры), которые вместо этого становятся синими, низкий рост , подвижность суставов , потерю слуха , проблемы с дыханием. [10] и проблемы с зубами ( несовершенный дентиногенез ). [5] Потенциально опасные для жизни осложнения , которые становятся более распространенными при более тяжелом ОИ, включают: разрыв ( расслоение ) крупных артерий , таких как аорта ; [1] : 333 [11] недостаточность легочного клапана вследствие деформации грудной клетки ; [1] : 335–341 [12] и базилярная инвагинация . [13] : 106–107
Основной механизм обычно заключается в проблеме соединительной ткани из-за отсутствия или плохого формирования коллагена I типа . [5] : 1513 Более чем в 90% случаев НО возникает вследствие мутаций генов COL1A1 или COL1A2 . [14] Эти мутации могут передаваться по наследству по аутосомно-доминантному типу, но могут также возникать спонтанно ( de novo ). [9] [15] Существует четыре клинически определяемых типа: тип I, наименее тяжелый; тип IV, средней степени тяжести; тип III, тяжелый и прогрессивно деформирующий; и тип II, перинатально летальный. [9] По состоянию на сентябрь 2021 г. [update]Известно, что 19 различных генов вызывают 21 документально подтвержденный генетически определенный тип ОИ, многие из которых чрезвычайно редки и зарегистрированы лишь у нескольких человек. [16] [17] Диагноз часто основывается на симптомах и может быть подтвержден биопсией коллагена или секвенированием ДНК . [10]
Хотя лекарства нет, [10] большинство случаев ОИ не оказывают существенного влияния на продолжительность жизни, [1] : 461 [15] смерть в детстве от него редка, [10] и многие взрослые с НО могут достичь значительной степени автономии , несмотря на инвалидность . [18] Поддержание здорового образа жизни путем физических упражнений , сбалансированного питания, достаточного с содержанием витамина D и кальция , а также отказ от курения могут помочь предотвратить переломы. [19] Люди с ОИ могут обратиться за генетической консультацией , чтобы предотвратить унаследование этого заболевания от них своими детьми. [1] : 101 Лечение может включать неотложную помощь при переломах костей, прием обезболивающих , физиотерапию , использование вспомогательных средств для передвижения, таких как фиксаторы для ног и инвалидные коляски . [10] прием добавок витамина D и, особенно в детстве, хирургическое вмешательство . [20] Роддинг — это имплантация металлических интрамедуллярных стержней вдоль длинных костей (например, бедренной кости ) с целью их укрепления. [10] Медицинские исследования также поддерживают использование препаратов класса бисфосфонатов , таких как памидронат , для увеличения плотности костей . [21] Бисфосфонаты особенно эффективны у детей; [22] однако неясно, улучшают ли они качество жизни или снижают частоту переломов. [7]
ОИ поражает только одного из 15 000–20 000 человек, что делает его редким генетическим заболеванием . [8] Результаты зависят от генетической причины заболевания (его типа). Тип I (наименее тяжелый) является наиболее распространенным, другие типы составляют меньшинство случаев. [15] [23] [24] ОИ средней и тяжелой степени в первую очередь влияет на подвижность; если операция с применением стержня проводится в детстве, некоторые из людей с более тяжелыми типами ОИ могут обрести способность ходить. [25] Состояние описано с древней истории. [26] Латинский ; термин «несовершенный остеогенез» был придуман голландским анатомом Виллемом Вроликом в 1849 году в буквальном переводе это означает «несовершенное формирование кости». [26] [27] : 683
Признаки и симптомы
[ редактировать ]Ортопедический
[ редактировать ]
Основным симптомом несовершенного остеогенеза является хрупкость костей с низкой минеральной плотностью ; все типы ОИ имеют некоторое поражение костей. [5] При умеренном и особенно тяжелом ОИ длинные кости могут искривляться , иногда очень сильно. [28] Из-за слабости костей они легко ломаются : исследование эндокринного отделения Национального института детского здоровья в Карачи, Пакистан, выявило в среднем 5,8 переломов в год у нелеченных детей. [29] После полового созревания переломы обычно возникают гораздо реже , но снова начинают увеличиваться у женщин после менопаузы и у мужчин в возрасте от 60 до 80 лет. [1] : 486
Гипермобильность суставов также является частым признаком ОИ, предположительно потому, что пораженные гены такие же, как и те, которые вызывают некоторые типы синдрома Элерса-Данлоса . [5] : 1513 [примечание 1] [30] [31]
Отологический
[ редактировать ]К 50 годам около 50% взрослых с ОИ испытывают значительную потерю слуха , гораздо раньше, чем в общей популяции. [32] Потеря слуха при ОИ может быть связана или не связана с видимыми деформациями косточек и внутреннего уха . [33] Потеря слуха часто начинается на втором, третьем и четвертом десятилетиях жизни и может быть кондуктивной , нейросенсорной или комбинацией обоих («смешанной»). [34] Если потеря слуха не происходит к 50 годам, вероятность ее возникновения в последующие годы значительно снижается. [32] Смешанная тугоухость наиболее распространена среди лиц с ОИ всех возрастных групп, тогда как кондуктивная тугоухость чаще всего поражает пожилых людей, а нейросенсорная тугоухость чаще поражает детей. [35]
Потеря слуха, связанная с ОИ, хотя и относительно редка, также может начаться в детстве; в исследовании сорока пяти детей в возрасте от четырех до шестнадцати лет было обнаружено, что двое страдают от этого заболевания — в возрасте 11 и 15 лет. [36] В другом исследовании 2008 года был проверен слух у 41 человека с НО. Результаты показали, что 88% людей старше 20 лет имели ту или иную форму потери слуха, и только 38% людей моложе 20 лет. [37]
Потеря слуха наиболее распространена при ОИ I типа; реже встречается при типах III и IV. [1] : 294–296 Другие части внутреннего уха также могут поражаться НО. вызывая проблемы с балансом ; однако лишь небольшие исследования обнаружили связь между головокружением и ОИ. [1] : 308 НО может ухудшить результаты лечения, направленного на устранение потери слуха. [35]
Помимо связи НО с нейросенсорной тугоухостью, НО связан с рядом неврологических нарушений, обычно затрагивающих центральную нервную систему , вследствие деформаций окружающих его скелетных структур. Неврологические осложнения, особенно базилярная инвагинация , могут отрицательно повлиять на продолжительность жизни. При ОИ это чаще всего связано с миграцией зубов вверх , [38] [13] : 106–107 особенность позвонка С2 . Нейрохирургия может потребоваться для коррекции тяжелых отклонений, когда они угрожают жизни пациента или вызывают сильные страдания или невыносимый неврологический дефицит . [38] [13] : 106–107
Системный
[ редактировать ]Поскольку его биологические причины были более точно определены, стало более широко признано, что, хотя первичный болезненный процесс НО происходит в костях, наиболее распространенные типы НО , вызванные мутациями гена коллагена I типа, поражают практически все кости. человеческого тела органы в некотором роде. [15]
Коллаген типа I присутствует во всех системах кровообращения и дыхания : от желудочков сердца до сердечных клапанов , сосудистой сети , [1] : 329 это неотъемлемая часть ткани легких . соединительной [1] : 336 Таким образом, сердечно-сосудистые осложнения, в том числе аортальная недостаточность , аневризма аорты и расслоение артерий , иногда сочетаются с ОИ. [1] : 333 но не так часто, как они сочетаются с синдромом Марфана . [1] : 332
Респираторные заболевания являются основной причиной смертности при ОИ. [12] [1] : 335 Наиболее очевидным источником респираторных проблем при ОИ является легочная недостаточность, вызванная нарушениями архитектуры грудной стенки . [1] : 341 Однако инфекции дыхательных путей , такие как пневмония , также более смертельны среди людей с ОИ, чем среди населения в целом. [12] [39] было обнаружено, что у людей с более тяжелыми деформациями грудной клетки наблюдается более сильное ограничение легких . В небольшом исследовании 2012 года, в котором приняли участие 22 итальянских пациента с НО типа III и IV, а также 26 здоровых людей из контрольной группы , [12]
ОИ, особенно его тяжелая форма типа III, также оказывает воздействие на желудочно-кишечную систему. было обнаружено, что это связано с рецидивирующей болью в животе и хроническим запором . В двух исследованиях на пациентах, страдающих ОИ, [40] [41] Особенно часто наблюдаются хронические запоры. [1] : 377 Считается, что состояние усугубляется асимметричным тазом ( выпячиванием вертлужной впадины ). [1] : 377 [41] Запор, связанный с ОИ, особенно в детстве, может вызывать чувство сытости и связанный с ним отказ от еды, что приводит к недостаточности питания . [1] : 377
Классификация
[ редактировать ]В современном использовании существуют две системы типизации ОИ. Первая, созданная Дэвидом Силленсом в 1979 году, классифицирует пациентов на четыре типа, или синдрома , в соответствии с их клинической картиной , без учета генетической причины их заболевания. [42] : 114–115 [43] Вторая система расширяет модель Sillence, но присваивает новые пронумерованные типы генетически по мере их обнаружения. [44] [43] Таким образом, людей с НО можно охарактеризовать как имеющих как клинический , так и генетический тип, которые могут быть эквивалентными, а могут и не быть эквивалентными. [43]
Тип I является наиболее распространенным, и 90% случаев возникают в результате мутаций COL1A1 или COL1A2 . [9] Симптомы широко варьируются в зависимости от типа, а также варьируются от человека к человеку, даже в одной семье. [45]
По состоянию на 2021 год определен 21 вид ОИ: [16] [17]
| Четыре типа Силленса | ||||||
|---|---|---|---|---|---|---|
| Тип | Описание | Ген | МОЙ БОГ | Способ наследования | Заболеваемость | Определенный [примечание 2] |
| я | мягкий | Нулевой COL1A1 аллель | 166200 | аутосомно-доминантный, 34% новых [6] | 1 из 30 000 [46] | 1979 [43] |
| II | летальный исход в перинатальном периоде [5] : 1511 | КОЛ1А1 , КОЛ1А2 | 166210 | аутосомно-доминантный, ≈100% de novo [6] | 1 из 40 000 [23] до 1 на 100 000 [46] | |
| III | тяжелая, прогрессирующая и деформирующая | КОЛ1А1 , КОЛ1А2 | 259420 | аутосомно-доминантный, 85% новых [6] | 1 из 60 000 [46] | |
| IV | изменчивая и деформирующаяся, но обычно с нормальной склерой [1] : 294–296 [47] | КОЛ1А1 , КОЛ1А2 | 166220 | аутосомно-доминантный, 50% новых [6] | 1 из 30 000 [1] : 21 | |
| Генетически определенные типы | ||||||
| Тип | Ген | МОЙ БОГ | Способ наследования [примечание 3] | Определенный [примечание 2] | ||
| V | ИФТМ5 | 610967 | аутосомно-доминантный [48] [49] | 2000 [50] [примечание 4] | ||
| МЫ | СЕРПИНФ1 | 613982 | аутосомно-рецессивный [48] | 2002 | ||
| VII | CRTAP | 610682 | аутосомно-рецессивный [48] | 2006 | ||
| VIII | ПРОКАЗА 1 | 610915 | аутосомно-рецессивный | 2007 | ||
| IX | ППИБ | 259440 | аутосомно-рецессивный | 2009 | ||
| Х | СЕРПИНХ1 | 613848 | аутосомно-рецессивный | 2010 | ||
| XI | ФКБП10 | 610968 | аутосомно-рецессивный | 2010 | ||
| XII | СП7 | 613849 | аутосомно-рецессивный | 2010 | ||
| XIII | БМП1 | 614856 | аутосомно-рецессивный | 2012 | ||
| XIV | ТМЕМ38Б | 615066 | аутосомно-рецессивный | 2012 | ||
| XV | ВНТ1 | 615220 | аутосомно-рецессивный | 2013 | ||
| XVI | CREB3L1 | 616229 | аутосомно-рецессивный | 2013 | ||
| XVII | СПАРК | 616507 | аутосомно-рецессивный | 2015 | ||
| XVIII | ТЕНТ5А | 617952 | аутосомно-рецессивный | 2018 | ||
| XIX | МБТПС2 | 301014 | Х-сцепленный рецессивный | 2016 | ||
| ХХ | МЭСР | 618644 | аутосомно-рецессивный | 2019 | ||
| XXI | КДЕЛР2 | 619131 | аутосомно-рецессивный | 2020 | ||
Типы тишины
[ редактировать ]


Четыре типа Силленса имеют как клиническое, так и генетическое значение; приведенные ниже описания являются клиническими и могут быть применены к нескольким генетическим типам ОИ. Когда оно используется как для обозначения генетического, так и клинического типа, это указывает на то, что клинические симптомы действительно вызваны мутациями в генах COL1A1 или COL1A2 , которые наследуются по аутосомно-доминантному типу. [16]
Тип I
[ редактировать ]Коллаген нормального качества, но вырабатывается в недостаточном количестве. [5] : 1516 Кости ломаются легче, чем у населения в целом, но не так легко, как при более тяжелых формах ОИ; может быть сколиоз , хотя и легкий по сравнению с НО типа III и IV, с меньшим углом Кобба ; суставы могут расшатываться; могут проявляться синие склеры; возможна потеря слуха; [47] : Таблица 1 и может быть небольшое уменьшение высоты. Поскольку существуют случаи отсутствия одного или нескольких из этих симптомов, ОИ типа I в некоторых случаях остается незамеченным во взрослом возрасте. [5] : 1513–1514
Некоторые далее разделяют тип I на типы I–A и I–B, которые определяются как отличающиеся отсутствием (I–A) или наличием (I–B) несовершенного дентиногенеза (опалесцирующих зубов). [47] [55] : 217 Люди с I типом обычно имеют нормальную продолжительность жизни. [56]
Тип II
[ редактировать ]Коллаген фатально дефектен на С-конце . [5] : 1512 В большинстве случаев смерть заканчивается вскоре после рождения или в течение первого года жизни из-за дыхательной недостаточности . Другой распространенной причиной смерти являются внутричерепные кровотечения из-за переломов черепа , возникших во время или вскоре после рождения. [5] : 1511 Во многих случаях новорожденный уже к моменту рождения имеет множественные переломы костей. У младенцев типа II также наблюдаются серьезные проблемы с дыханием и сильно деформируются кости. Шестьдесят процентов младенцев умирают менее чем через 24 часа после рождения, а выживание после первого года жизни крайне маловероятно и обычно требует искусственной вентиляции легких . [57] В редких случаях у младенцев, которые доживают до первого года жизни, серьезные задержки развития и моторики наблюдаются ; ни один из двух младенцев, исследованных в 2019 году, оба в возрасте около двух лет, не смогли контролировать голову , и обоим для дыхания требовался аппарат искусственной вентиляции легких. [58]
Тип II также известен как «летальная перинатальная » форма ОИ. [59] и несовместимо с выживанием во взрослой жизни. [57] Из-за столь же сильной деформации костей иногда младенцев с тяжелым типом III ошибочно первоначально относят к типу II; как только будет продемонстрирована долгосрочная выживаемость, вместо этого они считаются имеющими тип III. [5] : 1511 [60]
Тип III
[ редактировать ]Коллагена достаточно, но он недостаточно высокого качества. [5] : 1512 Клиническая дифференциация между типами III и IV не всегда проста и еще больше усложняется тем фактом, что у нелеченного взрослого человека с типом IV симптомы могут быть хуже, чем у взрослого человека с типом III, получающего лечение; [5] : 1511 [61] Особенностями, обнаруженными только у типа III, являются его прогрессивно деформирующийся характер. [5] : 1511–1512 и наличие лица «треугольной» внешности. [62] Еще одним отличительным фактором между типами III и IV являются голубые склеры; при типе III у младенцев обычно наблюдаются голубые склеры, которые с возрастом постепенно становятся белыми, но при типе IV синие склеры обычно не наблюдаются, [1] : 294–296 хотя они наблюдаются в 10% случаев. [63]
НО типа III вызывает остеопению костей, которые очень легко ломаются, иногда даже внутриутробно , что часто приводит к сотням переломов в течение жизни; [24] ранний сколиоз, прогрессирующий до полового созревания; карликовость (конечный рост взрослого человека часто менее 4 футов или 120 сантиметров); рыхлые суставы; и возможные проблемы с дыханием из-за малого объема грудной клетки , вызывающего малый объем легких . [5] : 1512
Из-за тяжести проблем с костями неврологические и судорожные расстройства . при III типе чаще развиваются [5] : 1512 Базилярная инвагинация , оказывающая давление на ствол мозга , может вызвать или способствовать ранней смерти; Хирургическое лечение в случаях ОИ более сложное. [5] : 1512 [13] : 106–107
Тип IV
[ редактировать ]Коллагена достаточно, но он недостаточно высокого качества. [5] : 1512 IV тип – для случаев различной степени тяжести, не вписывающихся ни в III, ни в I тип. [47] Хотя одной из необходимых характеристик Силленса для типа IV было наличие нормальных склер, [1] : 294–296 [42] : 114 Современная классификация позволяет даже людям с синими склерами соответствовать критериям типа IV, если они соответствуют другим клиническим требованиям этого типа. [63]
При типе IV деформация костей может быть от легкой до тяжелой, кости легко ломаются (особенно до полового созревания), часто наблюдается карликовость, очевиден коллапс позвонков и сколиоз, возможна потеря слуха. [10] хотя и редкость. [47] [46] ОИ типа IV в основном определяется в отличие от типа III и типа I, поскольку клиническая классификация пациентов находится где-то посередине между ними. [5] : 1511 Таким образом, НО типа IV часто называют «переменным» НО. [42] : 111 причем степень тяжести заболевания даже у членов одной семьи (то есть с одной и той же генетической мутацией) различается. [47]
Частота переломов костей в препубертатном возрасте является еще одним способом клинической оценки ОИ типа IV: у пациентов с ним частота переломов обычно составляет ≈1 в год по сравнению с ≈3 в год при тяжелом ОИ (тип III). [47]
Как и в случае с типом I, некоторые далее разделяют тип IV на типы IV-A и IV-B, определяемые также отсутствием (IV-A) или наличием (IV-B) несовершенного дентиногенеза . [47] [55] : 217
Генетически определенные типы (типы V – XXI )
[ редактировать ]По состоянию на 2020 год генетически определены пятнадцать типов ОИ: [16]
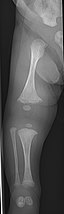


- Тип V. Имея те же клинические особенности, что и тип IV, его можно клинически отличить, наблюдая за «сетчатым» видом биопсии кости под микроскопом. Тип V можно отличить от других типов НО по «V-триаде»: непрозрачная полоса (видимая на рентгеновских снимках), примыкающая к пластинкам роста ; гипертрофические мозоли (аномально большие массы костной ткани), которые образуются в местах переломов в процессе заживления; и кальциноз межкостной перепонки предплечья , [50] что может затруднить поворот запястья. [1] : 429 Другие особенности этого состояния могут включать вытягивание локтя и, как и при других типах ОИ, искривление длинных костей и потерю слуха. [64] Случаи этого типа вызваны мутациями гена IFITM5 на хромосоме 11 p15.5. [64] [49] Отделение ОИ типа V от ОИ типа IV, его клинического типа, было первоначально предложено еще до того, как была известна его генетическая причина, Glorieux et al . в 2000 году. [50] [65] 4% пациентов с ОИ в генетическом отделении Бразильской клинической больницы Порту-Алегри . Тип V встречается относительно часто по сравнению с другими генетически определенными типами ОИ: он был обнаружен у [66]
- Тип VI . Обладая теми же клиническими особенностями, что и тип III, он отличается костями, которые имеют внешний вид, аналогичный тому, который наблюдается при остеомаляции . [1] : 168 Тип VI вызван мутацией потери функции гена SERPINF1 на хромосоме 17 p13.3. [1] : 170
- Тип VII – ОИ, вызванный мутацией гена CRTAP на хромосоме 3 p22.3; клинически сходен с ОИ типов II и III, в зависимости от конкретного человека. Тип VII был первым подтвержденным рецессивным типом ОИ, первоначально обнаруженным среди коренных народов Квебека . [67] [68]
- Тип VIII – ОИ, вызванный мутацией гена LEPRE1 на хромосоме 1 p34.2; клинически сходен с ОИ типов II и III, в зависимости от конкретного человека. [69]
- Тип IX – НО, вызванное гомозиготной или компаунд-гетерозиготной мутацией гена PPIB на хромосоме 15 q22.31. [70]
- Тип X – НО, вызванное гомозиготной мутацией гена SERPINH1 на хромосоме 11q13. [71]
- Тип XI – ОИ, вызванный мутациями FKBP10 на хромосоме 17q21. Мутации вызывают снижение секреции тримерных молекул проколлагена. Другие мутации в этом гене могут вызвать аутосомно-рецессивный синдром Брука , похожий на ОИ. [72]
- Тип XII – ОИ, вызванный мутацией сдвига рамки считывания SP7 на хромосоме 12 q13.13. Эта мутация вызывает деформации костей, переломы и задержку прорезывания зубов . [73]
- Тип XIII – НО, вызванный мутацией гена костного морфогенетического белка 1 ( BMP1 ) на хромосоме 8 p21.3. [74] Эта мутация вызывает повторные переломы, высокую костную массу и гипермобильность суставов . [75]
- Тип XIV – НО, вызванное мутациями гена TMEM38B на хромосоме 9 q31.2. Эта мутация вызывает рецидивирующие переломы и остеопению , хотя траектория заболевания весьма вариабельна. [76]
- Тип XV – НО, вызванное гомозиготными или компаунд-гетерозиготными мутациями гена WNT1 на хромосоме 12q13.12. Это аутосомно-рецессивный тип. [75]
- Тип XVI – НО, вызванный мутациями гена CREB3L1 на хромосоме 11p11.2. Гомозиготная мутация вызывает внутриутробное возникновение рецидивирующих переломов ребер и длинных костей, деминерализацию, снижение окостенения черепа и посинение склер; клинически это тип II или тип III. [77] Члены семьи, гетерозиготные по OI XVI, могут иметь рецидивирующие переломы, остеопению и синие склеры. [77] [78]
- Тип XVII – ОИ, вызванный гомозиготной мутацией гена SPARC на хромосоме 5 q33, вызывающей дефект белка остеонектина , что приводит к тяжелому заболеванию, характеризующемуся генерализованной платиспондилией , зависимостью от инвалидной коляски и рецидивирующими переломами. [79]
- Тип XVIII – НО, вызванное гомозиготной мутацией гена FAM46A на хромосоме 6 q14.1. Характеризуется врожденным искривлением длинных костей, червячными костями , синими склерами, коллапсом позвонков и множественными переломами в первые годы жизни. [80]
- Тип XIX – НО, вызванное гемизиготной мутацией гена MBTPS2 на Х-хромосоме p22.12. На сегодняшний день НО типа XIX является единственным известным типом НО с Х-сцепленным рецессивным типом наследования, что делает его единственным типом, который чаще встречается у мужчин, чем у женщин. НО типа XIX нарушает регулируемый внутримембранный протеолиз , который имеет решающее значение для формирования здоровой кости. [81]
- Тип XX – НО, вызванный гомозиготной мутацией гена MESD на хромосоме 15q25.1. Первоначальные исследования типа XX показывают, что он может вызывать глобальную задержку развития , впервые среди типов НО. НО типа XX нарушает сигнальный путь Wnt , который, как полагают, играет роль в развитии костей. [82]
- Тип XXI – НО, вызванное гомозиготной мутацией гена KDELR2 на хромосоме 7 p22.1. Вызывает заболевание, клинически сходное с типами II и III, которое, как полагают, связано с неспособностью белка-шаперона HP47 отсоединиться от коллагена типа I, поскольку для этого ему необходимо связываться с недостающим белком просвета ЭР, сохраняющим белок рецептора 2, кодируемый KDELR2 . [17]
Учитывая быструю скорость открытия типов, весьма вероятно, что существуют и другие гены, связанные с НО, о которых еще предстоит сообщить. [83] : 491–492
Генетика
[ редактировать ]
Несовершенный остеогенез — это группа генетических нарушений, каждое из которых приводит к хрупкости костей. НО имеет высокую генетическую гетерогенность , то есть множество различных генетических мутаций приводят к одинаковым или сходным наборам наблюдаемых симптомов ( фенотипов ). [84]
Основными причинами развития заболевания являются мутации в генах COL1A1 и/или COL1A2, которые совместно отвечают за выработку коллагена I типа . [85] Примерно 90% людей с НО являются гетерозиготными по мутациям в генах COL1A1 или COL1A2 . [86] Существует несколько биологических факторов, являющихся результатом доминирующей формы ОИ. Эти факторы включают: внутриклеточный стресс; аномальная минерализация тканей; аномальные межклеточные взаимодействия; аномальные клеток и матрикса взаимодействия ; нарушенная структура клеточного матрикса; и аномальное взаимодействие между неколлагеновыми белками и коллагеном. [87]
Предыдущие исследования привели к убеждению, что ОИ является аутосомно-доминантным заболеванием с небольшим количеством других вариантов генома. [8] Однако со снижением стоимости секвенирования ДНК в результате проекта «Геном человека» 2003 года были выявлены аутосомно-рецессивные формы заболевания. [88] Рецессивные формы НО в значительной степени связаны с дефектами шаперонов коллагена, ответственных за выработку проколлагена и сборку связанных с ним белков. [89] Примеры коллагеновых шаперонов , дефектных у пациентов с рецессивными формами ОИ, включают шаперон HSP47 ( синдром Коула-Карпентера ) и FKBP65 . [44] Мутации в этих шаперонах приводят к неправильному паттерну сворачивания белков коллагена 1, что вызывает рецессивную форму заболевания. [44] Выделяют три значимых типа ОИ, которые являются результатом мутаций в комплексе 3-гидроксилирования пролила коллагена (компоненты CRTAP, P3H1 и CyPB). [44] Эти компоненты ответственны за модификацию коллагена α1(l)Pro986. [44] Мутации в других генах, таких как SP7 , SERPINF1 , TMEM38B и BMP1, также могут приводить к неправильному формированию белков и ферментов , что приводит к другим рецессивным типам несовершенного остеогенеза. [44]
Дефекты белков фактора пигментного эпителия (PEDF) и костно-ограниченного интерферон-индуцированного трансмембранного белка (BRIL) являются причинами несовершенного остеогенеза V и VI типов. [90] Дефекты этих белков приводят к нарушению минерализации костей , что приводит к характерной хрупкости костей при несовершенном остеогенезе. [90] Единственная точечная мутация в 5'-нетранслируемой области (5'-UTR) гена IFITM5 , который кодирует BRIL, напрямую связана с ОИ типа V. [64] [91]
В редком случае типа XIX, впервые обнаруженном в 2016 году, НО наследуется как Х-сцепленное генетическое заболевание , пагубные последствия которого в конечном итоге возникают в результате мутации гена MBTPS2 . [81] Генетические исследования продолжаются, и неизвестно, когда будут идентифицированы все генетические причины ОИ, поскольку число генов, которые необходимо проверить, чтобы исключить заболевание, продолжает увеличиваться. [83] : 491–492
В исследовании 37 семей была обнаружена вероятность рецидива ОИ в 1,3% у нескольких братьев и сестер, рожденных от двух незатронутых родителей — это гораздо более высокий показатель, чем можно было бы ожидать, если бы все такие рецидивы возникали de novo . [92] Причина — генетический мозаицизм ; то есть некоторые или большая часть зародышевых клеток недостаточны одного родителя имеют доминантную форму ОИ, но их соматические клетки для того, чтобы вызвать симптомы или очевидную инвалидность у родителя - разные клетки родителя имеют две (или более ) наборы немного отличающихся ДНК. [92] [5] : 1513 Клинически отмечено, что ≈5–10% случаев НО II и III типов обусловлены генетическим мозаицизмом. [1] : 532
Патофизиология
[ редактировать ]Люди с НО либо рождаются с дефектной соединительной тканью , рождаются без способности производить ее в достаточных количествах, либо, в самых редких генетических типах, рождаются с недостатками других аспектов формирования кости , таких как белки-шапероны , сигнальный путь Wnt , BRIL и так далее. Белок [16] При I типе сама структура коллагена нормальная, просто его количество низкое. [5] : 1516 Типы II, III и IV обычно, но не всегда, связаны с дефицитом коллагена I типа . [93] Один из возможных дефицитов возникает из-за аминокислоты замены глицина на более объемную аминокислоту, такую как аланин , в коллагена белка тройной спиральной структуре . Более крупные боковые цепи аминокислот приводят к стерическим эффектам , которые создают выпуклость в коллагеновом комплексе , что, в свою очередь, влияет как на молекулярную наномеханику , так и на взаимодействие между молекулами , которые нарушаются. [94] [95] В зависимости от места замены и используемой вместо нее аминокислоты наблюдаются разные эффекты, которые объясняют разнообразие типов ОИ, несмотря на то, что в большинстве случаев ответственны одни и те же два гена коллагена. [96] [95] Замены глицина на серин или цистеин наблюдаются реже при фатальном ОИ II типа, тогда как замены на валин , аспарагиновую кислоту , глутаминовую кислоту или аргинин наблюдаются чаще. [95]
В более широком масштабе взаимоотношения между фибриллами коллагена и кристаллами гидроксиапатита , образующими кость, изменяются, вызывая хрупкость. [96] Переломы костей происходят из-за того, что стрессовое состояние внутри коллагеновых фибрилл изменяется в местах мутаций, где локально большие силы сдвига приводят к быстрому разрушению фибрилл даже при умеренных нагрузках, поскольку однородное стрессовое состояние, обычно встречающееся в здоровых коллагеновых фибриллах, теряется. [94] Таким образом, НО представляет собой многомасштабное явление, при котором дефекты на мельчайших уровнях тканей (генетическом, нано, микро) по принципу домино влияют на макроуровень тканей . [94]
Диагностика
[ редактировать ]
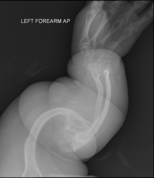
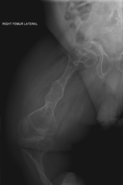
Диагноз обычно основывается на медицинских визуализациях , включая обычные рентгеновские снимки , и симптомах. При тяжелом ОИ признаки при медицинской визуализации включают изменения во всех конечностях и позвоночнике. [97] Поскольку рентгеновские лучи часто нечувствительны к сравнительно меньшей потере плотности костной ткани, связанной с ОИ типа I, сканирование DEXA . может потребоваться [5] : 1514
Диагноз ОИ может быть подтвержден с помощью анализа ДНК или коллагенового белка возникновения переломов костей при небольшой травме и наличия других клинических признаков, таких как голубые склеры , но во многих случаях для постановки диагноза достаточно . Биопсия кожи может быть выполнена для определения структуры и количества коллагена I типа. Хотя тестирование ДНК может подтвердить диагноз, оно не может его полностью исключить, поскольку не все мутации, вызывающие ОИ, еще известны и/или проверены. [83] : 491–492 НО II типа часто диагностируется с помощью УЗИ во время беременности, при котором уже могут быть видны множественные переломы и другие характерные признаки. По сравнению с контролем, OI кортикальной кости показывает повышенную пористость, диаметр канала и связность по данным микрокомпьютерной томографии. [98] ОИ также можно обнаружить до рождения с помощью метода генетического тестирования in vitro, такого как амниоцентрез . [99]
Генетическое тестирование
[ редактировать ]Чтобы определить наличие несовершенного остеогенеза, генетическое секвенирование наиболее распространенных проблемных генов: COL1A1 , COL1A2 и IFITM5 ; можно провести [100] если мутация не обнаружена, но подозрение на НО все еще сохраняется, можно протестировать еще 10+ генов, которые, как известно, вызывают НО. [16] Тестирование на дублирование и удаление также рекомендуется родителям, которые подозревают, что у их ребенка ОИ. [100] Наличие мутаций сдвига рамки считывания , вызванных дупликациями и делециями, обычно является причиной повышенной тяжести заболевания. [100]
Дифференциальный диагноз
[ редактировать ]Важным дифференциальным диагнозом ОИ является жестокое обращение с детьми , поскольку оба заболевания могут быть представлены врачу с множественными переломами на различных стадиях заживления. [5] : 1514 Дифференцировать их может быть сложно, особенно при отсутствии других характерных признаков ОИ. [1] : 391 Это может стать проблемой в суде; в Соединенных Штатах несколько дел о жестоком обращении с детьми были решены, и было установлено, что истинной причиной переломов ребенка был несовершенный остеогенез, что привело к судебным искам с требованием возмещения ущерба, таким как Алиса Веласкес и др. против Соединенных Штатов . [1] : 391 [101]
Другие дифференциальные диагнозы включают рахит и остеомаляцию , вызванные недостаточностью питания , а также редкие скелетные синдромы, такие как синдром Брука , гипофосфатазия , остеодиспластическая геродермия и синдром Элерса-Данлоса . [5] : 1513 [1] : 253–256 различные формы остеопороза , такие как ятрогенный остеопороз, идиопатический ювенильный остеопороз , остеопороз при неиспользовании и остеопороз, связанный с физической нагрузкой. При подозрении на ОИ также следует рассматривать [1] : 255–256
Уход
[ редактировать ]Лечения несовершенного остеогенеза не существует. [10] Поддержание здорового образа жизни путем физических упражнений и отказ от курения может помочь предотвратить переломы. [102] Лечение может включать уход за сломанными костями, прием обезболивающих, физиотерапию , использование вспомогательных средств для передвижения, таких как брекеты или инвалидные коляски, а также хирургическое вмешательство. [102]
Судить об успехе или неудаче лечения у пациентов с ОИ может быть сложно, поскольку снижение частоты переломов костей может быть просто случайным. Хотя эти показатели часто используются в медицинских исследованиях для оценки эффективности лечения, норвежское исследование пятнадцати человек с ОИ подчеркнуло, что, по их мнению, врачи должны учитывать всего пациента, а не только частоту переломов. [103]
Лечение острых переломов костей
[ редактировать ]Переломы костей лечат у людей с несовершенным остеогенезом во многом так же, как их лечат в общей популяции: кость с ОИ заживает с той же скоростью, что и кость без ОИ. [1] : 431 Больший упор делается на использование легких материалов для иммобилизации перелома, так как при умеренных или тяжелых типах ОИ использование тяжелых гипсовых повязок , таких как гипсовая повязка бедра, может вызвать переломы костей по границам гипсовой повязки, а также генерализованные остеопения . [1] : 431 Легкую гипсовую повязку или шину затем заменяют съемным ортезом через несколько недель, как только на рентгенограмме будут видны признаки сращения. [1] : 431 Во избежание несращения или неправильного сращения все переломы следует иммобилизировать, даже если перелом кажется незначительным ( микроперелом ), [1] : 439 поскольку люди с НО подвергаются большему риску несращения. [1] : 438
костей Инфекции , вторичные по отношению к переломам, лечатся по мере их возникновения соответствующими антибиотиками и антисептиками , как и в общей популяции. [1] : 424
Лекарства
[ редактировать ]Бисфосфонаты
[ редактировать ]
В 1998 году первоначальное обсервационное исследование продемонстрировало эффективность внутривенного введения памидроната , бисфосфоната , который ранее использовался у взрослых для лечения остеопороза . Это исследование показало, что при тяжелом ОИ памидронат уменьшает боль в костях, предотвращает новые переломы позвонков, изменяет форму ранее сломанных тел позвонков и снижает количество переломов длинных костей. [104]
Хотя пероральные бисфосфонаты удобнее и дешевле, они также плохо всасываются, а внутривенные бисфосфонаты, как правило, более эффективны, хотя этот вопрос находится в стадии изучения. Некоторые исследования показали, что пероральные и внутривенные бисфосфонаты, такие как пероральный алендронат и внутривенный памидронат, эквивалентны. [105] В двойном слепом исследовании 2013 года с участием детей с легкой формой ОИ пероральный прием ризедроната увеличивал минеральную плотность костей и уменьшал количество внепозвоночных переломов. Однако это не уменьшило количество новых переломов позвонков. [106] [107] Кокрейновский 2016 года пришел к выводу, что, хотя бисфосфонаты, по-видимому , обзор улучшают минеральную плотность костей, неясно, приводит ли это либо к снижению частоты переломов костей , либо к улучшению качества жизни людей с несовершенным остеогенезом. [7] Даже в исследованиях с участием 125 детей не было обнаружено причинно-следственной связи между бисфосфонатами и снижением частоты переломов; Плацебо-контролируемые исследования также не смогли доказать, что они приводят к увеличению силы, моторному контролю или снижению уровня боли. [87]
Бисфосфонаты не столь эффективны для увеличения минеральной плотности костей у взрослых. [22]
Пищевые добавки
[ редактировать ]НО является генетическим заболеванием и не вызван недостаточным потреблением каких-либо витаминов или минералов; добавки не могут вылечить ОИ. Тем не менее, люди с НО, как правило, испытывают серьезный дефицит витамина D, причем гораздо чаще, чем население в целом, и причина этого не совсем понятна. [108] [109] [110] Считается, что тяжесть дефицита и вероятность его возникновения связаны с тяжестью ОИ. [109] Могут быть рекомендованы добавки витамина D, по крайней мере, до тех пор, пока уровень 25(OH)D3 в крови пациента не вернется к норме. [108] Дефицит витамина D также вызывает беспокойство, поскольку он может снизить пользу от бисфосфанатов . [108]
Операция
[ редактировать ]Операция любого типа по своей сути несет в себе больший риск, если она проводится у пациента с ОИ (особенно от умеренной до тяжелой степени). Деформации скелета и несовершенный дентиногенез могут препятствовать доступу к дыхательным путям . [1] : 333 Использование и отказ от нее искусственной вентиляции легких также является более сложной задачей для пациентов с ОИ. [1] : 333 Во время самой процедуры или процесса заживления дефектный коллаген ОИ может привести к кровоточащим диатезам . [1] : 333
Безопасность анестезии также вызывает большее беспокойство у пациентов с ОИ. [1] : 333 вероятность возникновения анестезиологических осложнений в 5,6 раза выше при ОИ III типа. [111] Уникальной проблемой анестезии при ОИ являются периоперационные переломы - переломы, полученные в результате транспортировки пациента и методов доступа к дыхательным путям, которые, хотя и являются рутинными, когда кости пациента крепкие, могут привести к травмам из-за хрупкости костей ОИ. [112] Например, из-за сообщения 1972 года о плечевой кости переломе из-за манжеты сфигмоманометра , полученном у пациента с ОИ во время операции, протоколы мониторинга артериального давления часто модифицируются для пациентов с ОИ, при этом манжеты неонатального размера и настройки аппарата используются даже у взрослых; [113] : ¶11.72 кроме того, для наложения манжеты предпочтительно использовать наименее деформированную из конечностей пациента. [20] : ¶14.23
Роддинг
[ редактировать ]

Металлические стержни могут быть хирургическим путем вставлены в длинные кости для повышения прочности. Эта процедура была разработана Гарольдом А. Софилдом, когда он был руководителем аппарата детской больницы Шрайнерс в Чикаго , больницы, которая предлагает ортопедическую помощь и хирургические операции детям независимо от способностей их семьи. платить. [115] В Шрайнерс приходило большое количество детей с ОИ, и Софилд экспериментировал с различными методами укрепления их костей. [116] В 1959 году вместе с Эдвардом А. Милларом [ так в оригинале ] Софилд написал плодотворную статью, описывающую трехэтапную операцию, которая в то время казалась радикальной: точное разрушение костей («фрагментация»), размещение полученных костных фрагментов по прямой линии ( «перестройка»), а затем размещение металлических стержней в интрамедуллярных каналах длинных костей для их стабилизации и укрепления («фиксация стержней»). [117] Его лечение оказалось полезным для увеличения мобильности людей с ОИ, и оно было принято во всем мире — оно стало стандартным хирургическим лечением тяжелого ОИ к 1979 году, в этом году Дэвид Силленс обнаружил, что ≈ 2/3 обследованных им пациентов с НО типа III перенесли по крайней мере одну стержневую операцию. [42] : 108
Операцию Роддинга часто проводят в надежде, что она даст возможность передвигаться и ходить пациентам с умеренным или тяжелым ОИ. Обзор 2020 года, опубликованный в The Journal of Bone and Joint Surgery ( JB&JS ), показал, что он остается широко популярным: ≈ 2/3 людей с ОИ III и IV типов ( тяжелый ОИ) перенесли ту или иную форму стержневой хирургии в своей жизни, в среднем возрасте 4 + 1/10 и 7 + 1 / 2 года соответственно; [25] : Таблица I Одним из возможных объяснений тенденции к более раннему вмешательству при типе III является то, что половина больных детей вообще не могла ходить без операции, поскольку их конечности были более искривлены, поэтому операцию пришлось искать раньше. [25]
У пациентов с ОИ III типа, перенесших стержневую операцию, у 79,5% были стержневые бедренные и голени обеих ног. [25] : Таблица I Наиболее распространенной формой используемых стержней являются интрамедуллярные (IM) стержни , некоторые из которых, такие как IM стержень Фасье-Дюваля, являются телескопическими , то есть они предназначены для роста по мере роста ребенка, в попытке избежать необходимости ревизионные операции. [118] Широко используются телескопические стержни IM. [119] а обычный стержень Фасье-Дюваля IM предназначен для стержня бедренной, большеберцовой и плечевой костей. [120] : 1 Операция предполагает перелом длинных костей от одной до трех (или более) костей. [119] : Рисунок 4 местах, затем фиксируя стержень рядом с костью, чтобы она оставалась прямой. [120] : 11
Телескопические стержни IM предназначены для роста вместе с бедренной и большеберцовой костью у развивающихся детей; хирурги предпочитают использовать нетелескопические IM-стержни, такие как Rush, для большеберцовой кости, которая сравнительно меньше растет - обзор JB&JS показал, что, хотя 69,7% бедренных костей были пролечены с помощью телескопических IM-стержней, только 36,9% голеней были обработаны. [25] : Таблица IV
Хотя в обзоре JB&JS удалось связать получение стержневой операции с большей мобильностью при всех типах ОИ, у пациентов с типом IV операция не снизила частоту переломов костей по сравнению с пациентами без стержня, в то время как у пациентов с типом IV со стержневыми большеберцовыми костями наблюдалось 0,93 перелома большеберцовой кости в год, у пациентов с естественными большеберцовыми костями - только 0,81. Однако у пациентов с III типом стержневая операция снизила среднее количество переломов большеберцовой кости в год с 0,84 до 0,57. [25] : Таблица V
Спинной
[ редактировать ]Спондилодез может быть выполнен либо в качестве профилактической меры, либо для коррекции существующего сколиоза , хотя присущая хрупкость костей ОИ делает эту операцию более сложной у пациентов с ОИ, чем у пациентов с подростковым идиопатическим сколиозом, но с нормальной плотностью кости. [121] Однако, несмотря на риски, три хирурга-ортопеда Nemours-DuPont , специализирующиеся на хирургическом вмешательстве при несовершенном остеогенезе, рекомендуют операцию, если искривление превышает 50° после того, как ребенок преодолел максимальную скорость роста , поскольку искривление позвоночника может продолжать ухудшаться даже во взрослом возрасте. . [13] : 104
Из-за связанного с этим риска те же хирурги рекомендуют операцию по поводу базилярных отпечатков и базилярных инвагинаций проводить только в том случае, если давление, оказываемое на спинной мозг и ствол головного мозга, вызывает реальные неврологические симптомы. [13] : 106–107 Если базилярная инвагинация стала симптоматической, только хирургическое вмешательство может остановить или обратить вспять прогрессирование неврологического дефицита . [1] : 345
Физиотерапия
[ редактировать ]Обычно рекомендуется физиотерапия , однако из-за вариабельности ОИ требуются индивидуальные протоколы. [1] : 378 Физиотерапия используется для укрепления мышц, улучшения подвижности , гибкости и поддержания веса , однако ее следует проводить осторожно, чтобы свести к минимуму риск перелома костей. [1] : 378 У людей с ОИ упражнения часто включают водную аэробику , легкие упражнения с отягощениями и ходьбу, если пациент может. [1] : 378 Однако даже пациентам с легкой формой ОИ контактные виды спорта , а также виды деятельности, которые могут привести к ненужной нагрузке на суставы, например прыжки , противопоказаны из-за рисков, которые они представляют. [1] : 378
Людям с более ограниченной подвижностью рекомендуется регулярно менять положение в течение дня; Людям, которые сидят в инвалидной коляске большую часть или весь день, рекомендуется выходить из нее каждые два часа в качестве упражнения, чтобы уменьшить скованность и предотвратить образование пролежней . [1] : 378
Лица с ОИ средней и тяжелой степени, которым требуются вспомогательные средства передвижения и адаптированные транспортные средства , сталкиваются со значительными препятствиями при доступе к бассейнам или тренажерным залам, доступным для инвалидных колясок : у них либо нет ни одного бассейна поблизости, ни средств, чтобы туда добраться. [1] : 378 Ожирение чаще встречается у лиц с тяжелой формой ОИ (особенно после 20 лет) и у некоторых может привести к дальнейшему снижению подвижности. [122] [1] : 371, 373
на наклонном столе Вибрацию всего тела также можно проводить для увеличения мобильности длительно иммобилизованных ( прикованных к постели ) пациентов с ОИ; по крайней мере в двух случаях это помогло прикованным к постели детям сидеть прямо. [123] [87]
Зубы
[ редактировать ]Более чем у каждого второго человека с ОИ также имеется несовершенный дентиногенез (НД) — врожденная аномалия формирования дентина , одного из четырех основных компонентов человеческого зуба . [124] Стоматологическое лечение может представлять собой проблему из-за различных деформаций скелета и зубов, вызванных ОИ. Дети с НО должны пройти осмотр у стоматолога, как только у них прорежутся зубы ; это может свести к минимуму потерю структуры зубов в результате аномального дентина, и их следует регулярно контролировать, чтобы сохранить зубы и здоровье полости рта. [124]
Многих людей с ОИ лечат бисфосфонатами, и существует несколько возможных осложнений, связанных с стоматологическими процедурами, например, медикаментозный остеонекроз челюсти (MRONJ). 2016 года не было обнаружено сообщений о связанном с бисфосфонатами MRONJ ни у детей, ни у взрослых с ОИ . Однако в Кокрейновском обзоре безопасности и эффективности бисфосфонатов при ОИ [7]
В разработке
[ редактировать ]Моноклональные антитела
[ редактировать ]Моноклональные антитела уже давно рассматриваются для лечения ОИ, но по состоянию на 2021 год такая терапия не была одобрена для лечения ОИ ни в Европейском Союзе, ни в США. Таким образом, неясно, являются ли они безопасными и эффективными. Среди моноклональных антител, которые были изучены, - ромосозумаб (мишень для склеростина , компания Amgen ), фрезолимумаб ( TGF-β , Sanofi ), блозозумаб (склеростин, Lilly ) и сетрусумаб (склеростин, начатый Novartis ). [125]
Сетрусумаб, ранее известный как BPS-804, представляет собой моноклональное антитело, нацеленное на склеростин , и изучалось при ОИ больше, чем любое другое. В организме склеростин связывается с рецепторами LRP5 и LRP6 , что приводит к ингибированию сигнального пути Wnt . Это снижает костеобразование и не является проблемой, если у человека здоровые кости. [126] Однако считается, что снижение концентрации склеростина в организме может привести к образованию большего количества костей, и это является предпосылкой того, почему моноклональные антитела, которые снижают концентрацию естественного склеростина, могут помочь укрепить кости ОИ. [127] Хотя сетрусумаб был впервые разработан в фармацевтической компании Novartis, Novartis в 2015 году продала свои права на патент на препарат компании Mereo Biopharma, которая продолжила его разработку совместно с Ultragenyx . [125] [128] В 2019 году Mereo объявила о завершении сбора данных для исследования фазы II-B сетрусумаба; исследование завершилось 12 ноября 2020 года. [125] [129] Несмотря на то, что данные исследования не показали улучшения плотности костной ткани при QCT , что является его основной целью, улучшения были отмечены при DXA- сканировании. [130] В пресс-релизе от сентября 2020 года компания Mereo заявила, что намерена провести исследование фазы III в 2021 году и получила от Управления по контролю за продуктами и лекарствами США (FDA) статус редкого детского заболевания (RPD). [131]
Ромосозумаб , который также представляет собой моноклональное антитело, нацеленное на склеростин, является одобренным препаратом в США и ЕС для лечения остеопороза . Аналитик фармацевтической промышленности Evercore отметил, что «это может подорвать экономику Ultragenyx будет «жизненно важно» сетрусумаба», поскольку ромосозумаб стоит дешевле, чем лекарство от редкого заболевания, утверждая, что для прибыли доказать, что его сетрусумаб полезен. более эффективен, чем ромосозумаб, при ОИ. [125] Клиническое исследование по оценке эффективности ромосозумаба при ОИ началось в сентябре 2020 года и по состоянию на сентябрь 2021 года продолжается. [132] Ultragenyx прогнозирует, что вторая/третья фаза испытаний сетрусумаба завершится в 2026 году. [133] [134]
Профилактика
[ редактировать ]
Как генетическое заболевание, основа профилактики несовершенного остеогенеза в двадцать первом веке основана, в первую очередь, на предотвращении рождения пораженных людей . Генетическое консультирование может помочь пациентам и их семьям определить, какие виды скрининга, если таковые имеются, подходят для их ситуации. Пациенты могут рассмотреть возможность преимплантационной генетической диагностики после экстракорпорального оплодотворения , чтобы выбрать оплодотворенные эмбрионы, которые не пострадали. [1] : 247–248 Распространенные мутации, вызывающие ОИ, можно обнаружить с помощью секвенирования экзома и полногеномного секвенирования . Если беременность уже наступила, можно провести процедуру амниоцентеза, чтобы проверить, не затронут ли плод. [1] : 247 Если это произойдет, семья должна решить, хотят ли они прервать беременность и попытаться еще раз, что поднимает вопросы медицинской этики и права женщины на выбор . [135] [136]
Без вмешательства пациенты с наиболее распространенными мутациями, вызывающими несовершенный остеогенез, имеют 50% вероятность на протяжении беременности передачи заболевания , поскольку эти мутации наследуются по аутосомно-доминантному типу менделевского типа наследования . [1] : 247 У людей с редкими аутосомно-рецессивными формами ОИ вероятность передачи заболевания составляет 25%. Генетическое тестирование больных членов семьи можно использовать для определения того, какой тип наследования применим. [1] : 101
Поскольку тип I ОИ может быть трудно обнаружить у новорожденного ребенка, можно проверить пуповинную кровь ребенка, чтобы определить, был ли он передан, если семья уже отказалась от более инвазивных методов генетического скрининга. [1] : 247 В более тяжелых случаях диагноз можно поставить с помощью УЗИ, особенно если НО уже возможен. [1] : 248 Этическая проблема, связанная с пренатальным скринингом на ОИ, часто возникает, когда родители спрашивают, насколько серьезно пострадает их ребенок — на такие вопросы пока трудно дать однозначный ответ. [1] : 382
Если у незатронутого человека уже был ребенок с ОИ, существует большая вероятность (хотя и весьма малая) того, что у его будущих детей будет ОИ из-за генетического мозаицизма . [92] [1] : 100, 1513
Критика пренатального скрининга на НО в отношении прав инвалидов , проводимая некоторыми специалистами по биоэтике и некоторыми пострадавшими лицами, отрицательно сравнивает его с евгеникой , причем даже те, кто не выступает против абортов, выступают против выборочных абортов на этическом основании, что их существование выдает веру в то, что жизнь таких людей с ОИ «менее достойны жизни [и] менее ценны». [1] : 388
Прогноз
[ редактировать ]Прогноз § несовершенного остеогенеза полностью зависит от его типа (см. Классификация ).
Ожидаемая продолжительность жизни
[ редактировать ]При легкой форме заболевания типа I ожидаемая продолжительность жизни пациентов близка к таковой у населения в целом. [1] : 461 Однако при типе II пациенты очень редко доживают до двухлетнего возраста и обычно умирают в первые недели жизни. [5] : 1511 Оценка продолжительности жизни пациентов с типами III и IV более сложна, поскольку выбор образа жизни может привести к фатальным травматическим повреждениям, которые в противном случае не произошли бы или не привели бы к летальному исходу в общей популяции. Продолжительность жизни при ОИ IV типа считается близкой к нормальной, но при III типе она ниже, чем в общей популяции. [48]
Исследование данных Национального регистра пациентов показало, что при всех типах ОИ смертность от всех причин была в три раза выше, что привело к потере около семи лет у женщин и девяти лет у мужчин. [15] Исследование 1996 года, опубликованное в Британском медицинском журнале, показало, что смертность при ОИ III типа значительно выше: многие пациенты умирают в возрасте 20, 30 и 40 лет; Кроме того, было обнаружено, что пациенты, дожившие до 10-летнего возраста, имеют более продолжительную продолжительность жизни, чем новорожденные. [137]
Мобильность
[ редактировать ]Людям с легким (тип I) НО во взрослом возрасте требуется мало адаптивного оборудования , хотя в младенчестве они достигают двигательных показателей со значительной задержкой по сравнению с населением в целом. [1] : 477
С помощью адаптивного оборудования, такого как костыли , моторизованные инвалидные коляски , шины , удлинители и/или модификации дома, многие люди с умеренным и тяжелым ОИ могут достичь или сохранить значительную степень независимости . [18] [1] : 488 Ожидается, что при лечении и физиотерапии максимальным уровнем мобильности будет ходьба без посторонней помощи для типа I, ходьба по дому или физкультура для типа III и ходьба по дому или по месту жительства для типа IV; из-за различий в ОИ у разных людей достигнутая мобильность варьируется и может быть ниже ожидаемого максимума. [1] : 476
Эпидемиология
[ редактировать ]В Соединенных Штатах частота несовершенного остеогенеза оценивается в один случай на 20 000 живорождений. [138] По оценкам, от 20 000 до 50 000 человек страдают от ОИ в Соединенных Штатах. [139]
Наиболее распространенными типами являются I, II, III и IV, остальные встречаются очень редко. [140] Тип I является наиболее распространенным и, как сообщается, встречается примерно в три раза чаще, чем тип II. Распространенность типов III и IV менее определена. [23] В исследовании 1989 года, проведенном в Дании, было обнаружено, что тип I составляет 71% случаев, тип II - 12% случаев, а остальные типы составляют остальные 17%. [15] По данным исследования, проведенного в Швеции в 2015 году, тип I встречался почти в шесть раз чаще, чем тип III, и почти в четыре раза чаще, чем тип IV. [24]
Большинство людей с НО получают его от родителей, но во многих случаях это совершенно новая ( de novo или «спорадическая») мутация в семье. Среди исследованных пациентов с выживаемыми типами ОИ чаще всего встречается ОИ III типа (85%), за ним следуют тип IV (50%) и тип I (34%). [6] : Таблица 1
В некоторых группах населения заболеваемость ОИ может быть выше, чем можно было бы ожидать, если в них число носителей рецессивных форм заболевания превышает среднее. [1] : 20–21 [141]
История
[ редактировать ]

Это состояние или его виды на протяжении многих лет и у разных народов имели разные названия; Однако с конца 20-го века « остеогенез » несовершенный стал наиболее широко распространенным названием этого заболевания. Среди некоторых наиболее распространенных альтернатив - « fragilitas ossium »; [2] «Синдром Экмана-Лобштейна» и «синдром Вролика», оба эпонима ; и, в просторечии , «болезнь хрупких костей». [1]
Самые ранние зарегистрированные случаи
[ редактировать ]ОИ был идентифицирован у древнеегипетского младенца , мумифицированного примерно в 1000 г. до н.э., который первоначально был отвергнут археологами как содержащий останки обезьяны . [142] [143] : 161 Скандинавский Ивар король Бескостный , живший ок. 800 г. н. э. , [144] предполагают, что у него тоже был ОИ. [145]
Николя де Мальбранш часто считается первым человеком, описавшим физические характеристики НО в своей книге 1688 года « В поисках истины» , в которой он описывает человека, у которого «кости сломаны в тех местах, где мог бы быть сломан убийца». "Всю жизнь. Однако его уверенное описание патологии расстройства , которое создает то, что он назвал « enfants monstrueux » («чудовищные дети»), с научной точки зрения недействительно — он писал, что это произошло из-за того, что мать до родов наблюдала за публичной казнью путем поломки колеса. . [27] : 683 [143] : 165–168 [146]
Самые ранние современные научные исследования ОИ начались в 1788 году Олофом Якобом Экманом, который описал состояние, которое он назвал « остеомаляцией врожденной », в своей докторской диссертации и упомянул случаи его возникновения, начиная с 1678 года, все в одной семье, через трех поколения. [147] В описании этого состояния Экманом упоминались карликовость, хрупкость костей и искривление длинных костей. [148] : 763 В 1831 году Эдмунд Аксман дал подробное описание этого заболевания у себя и двух своих братьев, первым упомянув синие склеры как характерный признак ОИ. [27] : 683 Жан Лобштейн впервые описал легкую форму заболевания, сегодня известную как тип I, в 1833 году, назвав ее « остеопсатироз идиопатический ». [3] : 347
Лишь в 1912 году потеря слуха была признана симптомом ОИ, впервые упомянутая в краткой статье английского врача Чарльза Аллена Адэр-Дайтона. [149] [143] : 168–169
Термина
[ редактировать ]

Виллем Вролик , голландский анатом , который также был куратором «Музея Вроликианума», который сделал его причастным ко многим образцам тел, имеющих врожденные дефекты , придумал термин «несовершенный остеогенез». [27] : 683 в своей двуязычной на латыни и голландском языках книге по тератологии человека и млекопитающих» , «Иллюстрации эмбриогенеза впервые опубликованной в 1849 году. [150]
Включено описание останков младенца, у которого был так называемый перинатально фатальный ОИ II типа. [3] : 347 [1] : 5 (что подтверждено повторным исследованием останков в 1998 году Балджетом и др. ). [151] Останки сначала были переданы отцу Вролика, который не мог в них разобраться. Вролик описал плохо минерализованные кости, искривленные длинные кости и переломы в различных стадиях заживления. [152] Вролик правильно определил, что то, что он назвал ОИ, у младенца было вызвано не вторичным рахитом , а врожденной аномалией, вызывающей первичную остеопению ; он предположил, что это произошло из-за отсутствия «внутренней порождающей энергии». [151]
О своей классификации
[ редактировать ]Классификация НО также развивалась по мере улучшения его научного понимания. До появления современного генетического тестирования ОИ классифицировали на две широкие группы: несовершенный остеогенез врожденный и несовершенный остеогенез — поздний разделение, впервые предложенное немецким врачом Э. Лузером в 1906 году. [2] [153] Congenita использовалась для описания современных клинических типов II, III и некоторых случаев IV, когда при рождении состояние было очевидным либо из-за искривления конечностей, либо из-за переломов, внутриутробно полученных . [42] Тарда использовалась для классификации современного ОИ типа I и некоторых случаев типа IV, когда присущая хрупкость костей становилась очевидной лишь спустя долгое время после рождения. [2] Идея о том, что эти «поздние» и «пренатальные» формы являются проявлениями одного и того же расстройства, впервые была предложена в 1897 году Мартином Бенно Шмидтом ; [154] к 1950-м годам этот факт был широко принят. [3] : 346
Между тем современная система четырех типов (I, II, III, IV) была представлена в статье Дэвида Силленса , Элисон Сенн и Дэвида Дэнкса в Журнале медицинской генетики в 1979 году. [42] [155] и с тех пор стали стандартными терминами среди врачей, пациентов и исследователей. [43] [47] [156] Современные генетические типы (с номерами больше IV) вошли в употребление, поскольку с момента открытия первой из них Роем Морелло и др. было обнаружено все больше и больше рецессивно наследуемых форм ОИ. в 2006 году. [46] [8] [67] В 2010 году Международная номенклатурная группа конституциональных нарушений скелета (INCDS) «освободила» типы Силленс от молекулярной референции , согласившись на их новую клиническую роль после того, что для них стало «неожиданным» увеличением числа генетических причин ОИ. [156] [47]
В статье для Ежегодного обзора генетики в 2012 году д-р. Питер Байерс и Шона М. Пайотт посетовали, как расширение числа типов за счет включения генетических типов создало систему, которая « росла, как Топси ». [83] : 492 Они предполагают, что действительно может оказаться невозможным создать систему, которая была бы полезна для клиницистов и точно описывала бы генетическую причину ОИ человека, при этом попытки всегда отдают приоритет одному использованию в ущерб другому. [83] : 492
Общество и культура
[ редактировать ]Было проведено множество медицинских исследований причин несовершенного остеогенеза, которые принесли пользу не только людям с ОИ, но и медицине в целом; За десять лет с 2006 по 2016 год многочисленные открытия рецессивных генных мутаций, не связанных с коллагеном, которые до сих пор приводят к клиническим признакам ОИ у тех, у кого они есть, привели к многочисленным прорывам в медицинском понимании процесса здорового развития костей. . [8]
Другие животные
[ редактировать ]У собак ОИ является аутосомно-рецессивным заболеванием, то есть страдают собаки с двумя копиями аллеля. [157] Многие племенные организации и ветеринары предлагают тесты на НО, чтобы определить, является ли собака носителем НО. [157] [158] Чтобы предотвратить ОИ, собак, гетерозиготных по ОИ, следует сводить только с неносителями. [158]
Естественные мутации, вызывающие ОИ, были обнаружены у золотистых ретриверов , такс и биглей . НО также был выявлен у рыбок данио и мышей . [159]
Хотя собаки, мыши, рыбы и люди генетически не идентичны, некоторые из этих животных моделей официально признано, что представляют различные типы ОИ у людей. Например, у гомозиготных мышей oim / oim наблюдаются спонтанные переломы костей, небольшой размер тела и кифоз, что делает их моделью НО типа III. При этом гетерозиготная oim /+ мыши выглядят нормальными, но их кости немного слабее, чем у диких мышей, что делает их моделью ОИ типа I. [159] [160] Как и при НО человека, расположение мутировавшего гена влияет на тяжесть возникающего заболевания: мышь G859C Col1a1 является моделью НО типа II, поскольку все пораженные мыши умирают в перинатальном периоде. [159]
Тестирование на животных на идентифицированных моделях животных может привести к разработке лечения ОИ у людей. [159]
Пояснительные примечания
[ редактировать ]- ^ Одна мутация OI также вызывает комбинированный синдром Элера-Данлоса: «OIEDS1». ( Кабрал и др., 2005 г. , Расмуссен, 2020 г. )
- ^ Jump up to: а б Если год не указан, источником является OMIM.
- ^ Ссылки на гены для типов ОИ VII+ см. в записи о типах в § Генетически определяемые типы .
- ^ Генетическая причина не определена до 2012 года ( Cho, et al. 2012 ).
Ссылки
[ редактировать ]- ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С аа аб и объявление но из в ах есть также и аль являюсь а к ап ак с как в В из хорошо топор является тот нет бб до нашей эры др. быть парень бг чб с минет БК с бм млрд Шапиро-младший, Байерс П.Х., Глориё Ф.Х., Спонселлер П.Д. (2014). Несовершенный остеогенез: трансляционный подход к заболеванию хрупкой кости (2-е изд.). Лондон: Elsevier Inc. ISBN 978-0-12-397165-4 . OCLC 876364090 .
- ^ Jump up to: а б с д Роджер С., Фрэнсис М.Дж., Хоутон Г.Р. (1983). Синдром хрупкой кости: несовершенный остеогенез . Лондон: Баттерворт-Хайнеманн . п. 4. ISBN 978-0-407-00211-1 . OCLC 9784850 .
- ^ Jump up to: а б с д и Давел Дж.Г., Фихардт Т., Ван дер Спей Д. (октябрь 1956 г.). «Несовершенный остеогенез» . Архив болезней в детстве . 31 (159): 346–353. дои : 10.1136/adc.31.159.346 . ЧВК 2011939 . ПМИД 13363481 .
- ^ Jump up to: а б
- «остеогенез» . Lexico Британский словарь английского языка . Издательство Оксфордского университета . Архивировано из оригинала 23 августа 2021 года. «несовершенный остеогенез» . Lexico UK English Dictionary Британский словарь английского языка . Издательство Оксфордского университета . Архивировано из оригинала 23 августа 2021 года.
- «остеогенез» . Словарь Merriam-Webster.com . Проверено 23 августа 2021 г. «несовершенный остеогенез» . Словарь Merriam-Webster.com . Проверено 23 августа 2021 г.
- ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С Роу Д.В. (2008). «Несовершенный остеогенез». Принципы биологии кости (3-е изд.). Амстердам: Эльзевир . стр. 1511–1531. ISBN 978-0-12-373884-4 . OCLC 267135745 . Архивировано из оригинала 20 сентября 2021 года . Проверено 15 августа 2021 г.
Несовершенный дентиногенез (НД) наиболее часто встречается при ОИ III и IV типов и в целом поражает около 15% пациентов с ОИ различных фенотипов.
- ^ Jump up to: а б с д и ж Житник Л., Маасалу К., Дуй Б.Х., Пашенко А., Хмызов С., Рейманн Е. и др. (март 2019 г.). «De novo и наследственные патогенные варианты несовершенного остеогенеза, связанного с коллагеном» . Молекулярная генетика и геномная медицина . 7 (3): е559. дои : 10.1002/mgg3.559 . ПМК 6418448 . ПМИД 30675999 .
- ^ Jump up to: а б с д Дван К., Филиппи К.А., Штайнер Р.Д., Базель Д. (октябрь 2016 г.). «Бисфосфонатная терапия при несовершенном остеогенезе» . Кокрановская база данных систематических обзоров . 2016 (10): CD005088. дои : 10.1002/14651858.CD005088.pub4 . ПМК 6611487 . ПМИД 27760454 .
- ^ Jump up to: а б с д и Форлино А., Марини Дж.К. (апрель 2016 г.). «Несовершенный остеогенез» . Ланцет . 387 (10028): 1657–1671. дои : 10.1016/S0140-6736(15)00728-X . ПМЦ 7384887 . ПМИД 26542481 .
- ^ Jump up to: а б с д «Несовершенный остеогенез» . Домашний справочник по генетике . Национальная медицинская библиотека США , Национальные институты здравоохранения . 18 августа 2020 года. Архивировано из оригинала 29 августа 2021 года . Проверено 15 августа 2021 г.
- ^ Jump up to: а б с д и ж г час Ли Б., Краков Д. (июль 2019 г.). «Обзор несовершенного остеогенеза» . Национальный институт артрита, скелетно-мышечных и кожных заболеваний . Национальные институты здравоохранения . Архивировано из оригинала 9 мая 2021 года . Проверено 21 августа 2021 г.
- ^ Макнили М.Ф., Дончос Б.Н., Лафламм М.А., Хубка М., Садро К.Т. (декабрь 2012 г.). «Расслоение аорты при несовершенном остеогенезе: описание случая и обзор литературы». Экстренная радиология . 19 (6): 553–556. дои : 10.1007/s10140-012-1044-1 . ПМИД 22527359 . S2CID 11109481 .
- ^ Jump up to: а б с д ЛоМауро А, Починтеста С, Ромей М, Д'Анджело МГ, Педотти А, Туркони АС, Аливерти А (27 апреля 2012 г.). «Деформации грудной клетки изменяют работу дыхательных мышц и функцию грудной клетки у пациентов с тяжелым несовершенным остеогенезом» . ПЛОС ОДИН . 7 (4): e35965. Бибкод : 2012PLoSO...735965L . дои : 10.1371/journal.pone.0035965 . ПМЦ 3338769 . ПМИД 22558284 .
- ^ Jump up to: а б с д и ж Уоллес М.Дж., Круз Р.В., Шах С.А. (февраль 2017 г.). «Позвоночник у пациентов с несовершенным остеогенезом». Журнал Американской академии хирургов-ортопедов . 25 (2): 100–109. дои : 10.5435/JAAOS-D-15-00169 . ПМИД 28009707 .
- ^ Шапиро-младший, спонсор PD (23 августа 2005 г.). «10 — Несовершенный остеогенез». В Kleerekoper M, Siris ES, McClung M (ред.). Руководство по костям и минералам: Практическое руководство . Эльзевир Инк. п. 45. ИСБН 978-0-08-047074-0 . Архивировано из оригинала 20 сентября 2021 года . Проверено 17 сентября 2021 г.
- ^ Jump up to: а б с д и ж Фолкестад Л., Хальд Дж.Д., Канудас-Ромо В., Грам Дж., Герман А.П., Лангдаль Б. и др. (декабрь 2016 г.). «Смертность и причины смерти у пациентов с несовершенным остеогенезом: общенациональное когортное исследование на основе регистров» . Журнал исследований костей и минералов . 31 (12): 2159–2166. дои : 10.1002/jbmr.2895 . ПМИД 27345018 . S2CID 32196304 .
- ^ Jump up to: а б с д и ж Маром Р., Рабенхорст Б.М., Морелло Р. (октябрь 2020 г.). «Несовершенный остеогенез: обновленная информация о клинических особенностях и методах лечения» . Европейский журнал эндокринологии . 183 (4): Р95–Р106. doi : 10.1530/EJE-20-0299 . ПМЦ 7694877 . ПМИД 32621590 . Архивировано из оригинала 17 августа 2021 года . Проверено 17 августа 2021 г.
- ^ Jump up to: а б с ван Дейк Ф.С., Семлер О., Этих Дж., Кёлер А., Хименес-Эстрада Х.А., Бравенбоер Н. и др. (ноябрь 2020 г.). «Взаимодействие между KDELR2 и HSP47 как ключевой детерминант несовершенного остеогенеза, вызванного биаллельными вариантами KDELR2» . Американский журнал генетики человека . 107 (5): 989–999. дои : 10.1016/j.ajhg.2020.09.009 . ПМЦ 7675035 . ПМИД 33053334 . Архивировано из оригинала 20 сентября 2021 года . Проверено 29 августа 2021 г.
- ^ Jump up to: а б Донохо М (2020). «Терапия, ортопедические изделия и вспомогательные устройства при несовершенном остеогенезе» . В Крузе RW (ред.). Несовершенный остеогенез . Международное издательство Спрингер. стр. 21–37. дои : 10.1007/978-3-030-42527-2_3 . ISBN 978-3-030-42526-5 . S2CID 219518604 .
{{cite book}}:|work=игнорируется ( помогите ) - ^ «Что нужно знать людям с несовершенным остеопорозом об остеопорозе» . Национальный ресурсный центр остеопороза и связанных с ним заболеваний костей . Национальные институты здравоохранения . Ноябрь 2018. Архивировано из оригинала 18 марта 2021 года . Проверено 18 сентября 2021 г.
- ^ Jump up to: а б Эспозито П., Уоллес М.Дж. (2020), Круз Р.В. (ред.), «Хирургическое лечение несовершенного остеогенеза бедренной кости и колена» (ePub) , Несовершенный остеогенез: руководство по принятию хирургических решений и уходу на основе конкретных случаев , Springer International Publishing , стр. 147–182, номер документа : 10.1007/978-3-030-42527-2_10 , ISBN. 978-3-030-42527-2 , S2CID 219515805
- ^ Харрингтон Дж., Сочетт Э., Ховард А. (декабрь 2014 г.). «Обновленная информация об оценке и лечении несовершенного остеогенеза». Детские клиники Северной Америки . 61 (6): 1243–1257. дои : 10.1016/j.pcl.2014.08.010 . ПМИД 25439022 .
- ^ Jump up to: а б Шеврел Г., Шотт А.М., Фонтанж Э., Шаррен Дж.Э., Лина-Гранад Г., Дюбеф Ф. и др. (февраль 2006 г.). «Влияние перорального алендроната на МПК у взрослых пациентов с несовершенным остеогенезом: 3-летнее рандомизированное плацебо-контролируемое исследование» . Журнал исследований костей и минералов . 21 (2): 300–306. дои : 10.1359/JBMR.051015 . ПМИД 16418786 . S2CID 34089615 .
- ^ Jump up to: а б с Субраманиан С., Вишванатан В.К. (2021). Несовершенный остеогенез . СтатПерлз. ПМИД 30725642 . Архивировано из оригинала 21 октября 2020 года . Проверено 30 июля 2019 г.
{{cite book}}:|website=игнорируется ( помощь ) Последнее обновление: 3 февраля 2019 г. - ^ Jump up to: а б с Линдал К., Острем Э., Рубин С.Дж., Григелиониене Г., Мальмгрен Б., Юнггрен О., Киндмарк А. (август 2015 г.). «Генетическая эпидемиология, распространенность и корреляции генотип-фенотип в шведской популяции с несовершенным остеогенезом» . Европейский журнал генетики человека . 23 (8): 1042–1050. дои : 10.1038/ejhg.2015.81 . ПМЦ 4795106 . ПМИД 25944380 .
- ^ Jump up to: а б с д и ж Родригес Селин М., Крюгер К.М., Каудилл А., Нагамани СК, Харрис Г.Ф., Смит П.А. (11 сентября 2020 г.). «Многоцентровое исследование интрамедуллярного стержня при несовершенном остеогенезе» . JB и JS в открытом доступе . 5 (3): е20.00031. дои : 10.2106/JBJS.OA.20.00031 . ПМЦ 7489747 . ПМИД 32984750 .
- ^ Jump up to: а б Келли Э.Б. (2012). Энциклопедия генетики человека и болезней . АВС-КЛИО. п. 613. ИСБН 9780313387135 . Архивировано из оригинала 5 ноября 2017 года.
- ^ Jump up to: а б с д и ж Данг До АН, Марини Дж.К. (27 января 2021 г.). «Несовершенный остеогенез» . Кэри Дж.К., Кэссиди С.Б., Батталья А., Вискочил Д. (ред.). Управление генетическими синдромами Кэссиди и Аллансона . Джон Уайли и сыновья. стр. 683–705. ISBN 978-1-119-43267-8 . Архивировано из оригинала 29 августа 2021 года . Проверено 29 августа 2021 г.
- ^ Джонсон М.Т., Моррисон С., Хигер С., Муни С., Байерс П.Х., Робин Н.Х. (февраль 2002 г.). «Вариант несовершенного остеогенеза IV типа с разрешающейся кифомелией вызван новой мутацией COL1A2» . Журнал медицинской генетики . 39 (2): 128–132. дои : 10.1136/jmg.39.2.128 . ПМК 1735034 . ПМИД 11836364 .
- ^ Атта И., Икбал Ф., Лоун С.В. и др. (2014). «Эффект внутривенного лечения памидронатом у детей с несовершенным остеогенезом» (PDF) . Журнал Колледжа врачей и хирургов Пакистана . 24 (9). Карачи: 653–657. ПМИД 25233970 . Архивировано (PDF) из оригинала 17 августа 2021 года . Проверено 12 декабря 2023 г.
- ^ Кабрал В.А., Макареева Е., Колидж А., Леточа А.Д., Тай Дж.М., Йоуэлл Х.Н. и др. (май 2005 г.). «Мутации вблизи амино-конца коллагена альфа1 (I) вызывают комбинированный несовершенный остеогенез / синдром Элерса-Данлоса из-за вмешательства в процессинг N-пропептида» . Журнал биологической химии . 280 (19): 19259–19269. дои : 10.1074/jbc.m414698200 . ПМИД 15728585 . Архивировано из оригинала 20 сентября 2021 года . Проверено 17 августа 2021 г.
- ^ Расмуссен С.А. (2 декабря 2020 г.). «Комбинированный несовершенный остеогенез и синдром Элерса-Данлоса 1; OIEDS1» . Интернет-менделевское наследование у человека . Университет Джонса Хопкинса . 619115. Архивировано из оригинала 20 сентября 2021 года . Проверено 17 августа 2021 г.
- ^ Jump up to: а б Патерсон Ч.Р., Монк Э.А., Макаллион С.Дж. (апрель 2001 г.). «Насколько распространено нарушение слуха при несовершенном остеогенезе?» . Журнал ларингологии и отологии . 115 (4): 280–282. дои : 10.1258/0022215011907442 . ПМИД 11276328 . S2CID 12303464 . Архивировано из оригинала 20 сентября 2021 года . Проверено 15 августа 2021 г.
- ^ Верник Д. (2 ноября 2005 г.). «Проблемы ОИ: потеря слуха» . Архивировано из оригинала 20 января 2012 года . Проверено 4 ноября 2018 г.
- ^ Сантос Ф., МакКолл А.А., Чиен В., Мерчант С. (декабрь 2012 г.). «Отопатология при несовершенном остеогенезе» . Отология и невротология . 33 (9): 1562–1566. дои : 10.1097/МАО.0b013e31826bf19b . ПМЦ 3498599 . ПМИД 22996160 .
- ^ Jump up to: а б Пиллион Дж.П., Верник Д., Шапиро Дж. (14 декабря 2011 г.). «Потеря слуха при несовершенном остеогенезе: характеристики и рекомендации по лечению» . Международное генетическое исследование . 2011 . Хидави : 983942. doi : 10.4061/2011/983942 . ПМЦ 3335494 . ПМИД 22567374 .
- ^ Куурила К., Гренман Р., Йоханссон Р., Кайтила И. (июль 2000 г.). «Нарушение слуха у детей с несовершенным остеогенезом» . Европейский журнал педиатрии . 159 (7): 515–519. дои : 10.1007/s004310051322 . ПМИД 10923226 . S2CID 8729406 . Архивировано из оригинала 15 августа 2021 года . Проверено 15 августа 2021 г.
- ^ Пиллион Дж. П., Шапиро Дж. (сентябрь 2008 г.). «Аудиологические данные при несовершенном остеогенезе». Журнал Американской академии аудиологии . 19 (8): 595–601. дои : 10.3766/jaaa.19.8.3 . ПМИД 19323351 .
- ^ Jump up to: а б Ханданпур Н., Коннолли Д.Д., Рагхаван А., Гриффитс П.Д., Хоггард Н. (1 декабря 2012 г.). «Краниоспинальные аномалии и неврологические осложнения несовершенного остеогенеза: обзор изображений» . Рентгенография . 32 (7): 2101–2112. дои : 10.1148/rg.327125716 . ПМИД 23150860 . Архивировано из оригинала 18 августа 2021 года . Проверено 18 августа 2021 г.
- ^ Туркаль М., Миранович В., Лулич-Юревич Р., Гергья Юрашки Р., Приморац Д. (25 сентября 2017 г.). «Кардиореспираторные осложнения у больных с несовершенным остеогенезом» . Педиатрия Хорватии . 61 (3): 106–111. ISSN 1330-1403 . Архивировано из оригинала 20 сентября 2021 года . Проверено 29 августа 2021 г.
- ^ Виолас П., Фасье Ф., Хамди Р., Дюэм М., Глорьё Ф.Х. (сентябрь 2002 г.). «Выпячивание вертлужной впадины при несовершенном остеогенезе». Журнал детской ортопедии . 22 (5): 622–625. дои : 10.1097/01241398-200209000-00010 . ПМИД 12198464 . S2CID 37895736 .
- ^ Jump up to: а б Ли Дж.Х., Гэмбл Дж.Г., Мур Р.Э., Рински Л.А. (сентябрь 1995 г.). «Желудочно-кишечные проблемы у пациентов с несовершенным остеогенезом III типа». Журнал костной и суставной хирургии. Американский том . 77 (9): 1352–1356. дои : 10.2106/00004623-199509000-00010 . ПМИД 7673285 .
- ^ Jump up to: а б с д и ж Силленс Д.О., Сенн А., Дэнкс Д.М. (апрель 1979 г.). «Генетическая гетерогенность при несовершенном остеогенезе» . Журнал медицинской генетики . 16 (2): 101–116. дои : 10.1136/jmg.16.2.101 . ПМЦ 1012733 . ПМИД 458828 .
- ^ Jump up to: а б с д и Ван Дейк Ф.С., Палс Дж., Ван Рейн Р.Р., Никкельс П.Г., Коббен Дж.М. (1 января 2010 г.). «Пересмотр классификации несовершенного остеогенеза» . Европейский журнал медицинской генетики . 53 (1): 1–5. дои : 10.1016/j.ejmg.2009.10.007 . ПМИД 19878741 . Архивировано из оригинала 21 августа 2021 года . Проверено 21 августа 2021 г.
В 1979 году Силленс предложил классификацию несовершенного остеогенеза (НО) на типы НО I, II, III и IV. В 2004 и 2007 годах эта классификация была расширена за счет НО типов V–VIII из-за различных клинических особенностей и/или различных причинных мутаций генов.
- ^ Jump up to: а б с д и ж Марини Дж. К., Блиссетт А. Р. (август 2013 г.). «Новые гены в развитии костей: что нового в несовершенном остеогенезе» . Журнал клинической эндокринологии и метаболизма . 98 (8): 3095–3103. дои : 10.1210/jc.2013-1505 . ПМЦ 3733862 . ПМИД 23771926 .
- ^ МакКьюсик В.А., Хамош А. (23 октября 2018 г.). «Несовершенный остеогенез I типа; OI1» . Интернет-менделевское наследование у человека . Университет Джонса Хопкинса . 166200. Архивировано из оригинала 26 марта 2021 года . Проверено 17 августа 2021 г.
- ^ Jump up to: а б с д и ван Дейк Ф.С., Коббен Дж.М., Кариминежад А., Маугери А., Никкельс П.Г., ван Рейн Р.Р., Палс Г. (декабрь 2011 г.). «Несовершенный остеогенез: обзор с клиническими примерами» . Молекулярная синдромология . 2 (1): 1–20. дои : 10.1159/000332228 . ПМЦ 3343766 . ПМИД 22570641 .
- ^ Jump up to: а б с д и ж г час я дж Ван Дейк Ф.С., Силленс Д.О. (июнь 2014 г.). «Несовершенный остеогенез: клинический диагноз, номенклатура и оценка степени тяжести» . Американский журнал медицинской генетики. Часть А. 164А (6): 1470–1481. дои : 10.1002/ajmg.a.36545 . ПМЦ 4314691 . ПМИД 24715559 .
- ^ Jump up to: а б с д Штайнер Р.Д., Пепин М.Г., Байерс П.Х., Пагон Р.А., Берд Т.Д., Долан Ч.Р., Стивенс К., Адам М.П. (6 мая 2021 г.). « COL1A1/2 Несовершенный остеогенез». В Адаме М.П., Ардингере Х.Х., Пагоне Р.А. и др. (ред.). GeneReviews [Интернет] . Вашингтонский университет, Сиэтл. ПМИД 20301472 . Архивировано из оригинала 18 января 2017 года . Проверено 26 августа 2021 г.
- ^ Jump up to: а б Шапиро Дж.Р., Литман С., Гровер М., Лу Дж.Т., Нагамани С.К., Доусон BC и др. (июль 2013 г.). «Фенотипическая изменчивость несовершенного остеогенеза типа V, вызванная мутацией IFITM5» . Журнал исследований костей и минералов . 28 (7): 1523–1530. дои : 10.1002/jbmr.1891 . ПМЦ 3688672 . ПМИД 23408678 .
- ^ Jump up to: а б с Глориё Ф.Х., Раух Ф., Плоткин Х., Уорд Л., Трэверс Р., Раули П. и др. (сентябрь 2000 г.). «Несовершенный остеогенез типа V: новая форма болезни хрупких костей». Журнал исследований костей и минералов . 15 (9): 1650–1658. дои : 10.1359/jbmr.2000.15.9.1650 . ПМИД 10976985 . S2CID 13748803 .
- ^ Радд А. (26 марта 2016 г.). «Джордан Уийти: «Достичь совершенства невозможно. Ты должен быть тем, кто ты есть, потому что кто-то найдет тебя привлекательным» » . Таймс . ISSN 0140-0460 . Проверено 26 марта 2022 г.
- ^ Харви К. (6 марта 2015 г.). « Средняя звезда говорит, что быть ботаником — это нормально» . Вещи . Новая Зеландия: Stuff Ltd. Проверено 25 мая 2018 г.
- ^ «Актер и борец за права инвалидов: Питер Радтке умер» [Актер и борец за права инвалидов Питер Радтке умер]. Зеркало (на немецком языке). 30 ноября 2020 г. ISSN 2195-1349 . Проверено 26 марта 2022 г.
- ^ «Терезия Хайдлмайр» . Behinderte Menschen in Not (на австрийском немецком языке). Архивировано из оригинала 4 марта 2016 года . Проверено 26 марта 2022 г.
- ^ Jump up to: а б Певец Р.Б., Огстон С.А., Патерсон С.Р. (2001). «Смертность при различных типах несовершенного остеогенеза» . Журнал страховой медицины . 33 (3): 216–220. ПМИД 11558400 . Архивировано из оригинала 15 августа 2021 года . Проверено 15 августа 2021 г.
- ^ «Несовершенный остеогенез: типы, симптомы и лечение» . Кливлендская клиника . 5 мая 2021 года. Архивировано из оригинала 17 августа 2021 года . Проверено 17 августа 2021 г.
- ^ Jump up to: а б Базель Д., Штайнер Р.Д. (июнь 2009 г.). «Несовершенный остеогенез: недавние открытия проливают новый свет на это когда-то хорошо изученное состояние» (PDF) . Генетика в медицине . 11 (6): 375–385. дои : 10.1097/GIM.0b013e3181a1ff7b . ПМИД 19533842 . Архивировано (PDF) оригинала 20 сентября 2021 года . Проверено 19 августа 2021 г.
- ^ Надар Р., Сарафф В., Хоглер В., Десаи М., Шоу Н. (31 июля 2019 г.). «Детерминанты выживания при несовершенном остеогенезе (НО) типа II» . Рефераты по костям . 7:31 . doi : 10.1530/boneabs.7.P31 . ISSN 2052-1219 . S2CID 208412310 . Архивировано из оригинала 20 сентября 2021 года . Проверено 19 августа 2021 г.
- ^ Зиса Д., Шаббир А., Мастри М., Сузуки Г., Ли Т. (ноябрь 2009 г.). «Внутримышечный VEGF восстанавливает поврежденное сердце: роль факторов роста хозяина и мобилизация клеток-предшественников» . Американский журнал физиологии. Регуляторная, интегративная и сравнительная физиология . 297 (5): R1503–R1515. дои : 10.1152/ajpregu.00227.2009 . ПМЦ 2777764 . ПМИД 19759338 .
- ^ Бен Амор IM, Глориё Ф.Х., Раух Ф. (6 сентября 2011 г.). «Генотип-фенотипические корреляции при аутосомно-доминантном несовершенном остеогенезе» . Журнал остеопороза . 2011 : 540178. doi : 10.4061/2011/540178 . ПМК 3170785 . ПМИД 21912751 .
Например, может случиться так, что на пренатальном УЗИ у плода диагностируется ОИ типа II, летальная форма, но после рождения оказывается, что он поражен менее серьезно, чем первоначально предполагалось, и в конечном итоге переклассифицируется как ОИ типа III или даже типа VI. если адекватное лечение начато на ранней стадии.
- ^ Георгеску I, Влад С, Гавриу Т.С., Дэн С., Пырван А.А. (июнь 2013 г.). «Хирургическое лечение несовершенного остеогенеза – 10-летний опыт» . Журнал медицины и жизни . 6 (2): 205–213. ПМЦ 3725451 . ПМИД 23904885 .
- ^ Чен Х (2006). «Треугольное лицо» . Атлас генетической диагностики и консультирования . Тотова, Нью-Джерси: Humana Press. п. 771. ИСБН 978-1-58829-681-8 . Архивировано из оригинала 8 июня 2013 года.
- ^ Jump up to: а б Джонсон М.Т., Моррисон С., Хигер С., Муни С., Байерс П.Х., Робин Н.Х. (февраль 2002 г.). «Вариант несовершенного остеогенеза IV типа с разрешающейся кифомелией вызван новой мутацией COL1A2» . Журнал медицинской генетики . 39 (2): 128–132. дои : 10.1136/jmg.39.2.128 . ПМК 1735034 . ПМИД 11836364 .
Стойкая бледно-голубая склера редко встречается при ОИ IV типа, но может наблюдаться у 10% больных.
- ^ Jump up to: а б с Чо Т.Дж., Ли К.Е., Ли С.К., Сонг С.Дж., Ким К.Дж., Чон Д. и др. (август 2012 г.). «Одна рекуррентная мутация в 5'-UTR IFITM5 вызывает несовершенный остеогенез типа V» . Американский журнал генетики человека . 91 (2): 343–348. дои : 10.1016/j.ajhg.2012.06.005 . ПМЦ 3415533 . ПМИД 22863190 .
- ^ Цао Ю.Дж., Вэй Цз., Чжан Х., Чжан З.Л. (12 июня 2019 г.). «Расширение клинического спектра несовершенного остеогенеза типа V: 13 дополнительных пациентов и обзор» . Границы эндокринологии . 10 : 375. дои : 10.3389/fendo.2019.00375 . ПМК 6581704 . ПМИД 31244780 .
- ^ Бризола Э., Маттос Э.П., Феррари Дж., Фрейре П.О., Гермер Р., Ллерена Х.К., Феликс ТМ (октябрь 2015 г.). «Клиническая и молекулярная характеристика несовершенного остеогенеза типа V» . Молекулярная синдромология . 6 (4): 164–172. дои : 10.1159/000439506 . ПМЦ 4662268 . ПМИД 26648832 .
- ^ Jump up to: а б Морелло Р., Бертин Т.К., Чен Ю., Хикс Дж., Тоначини Л., Монтикон М. и др. (октябрь 2006 г.). «CRTAP необходим для пролил-3-гидроксилирования, а мутации вызывают несовершенный рецессивный остеогенез» . Клетка . 127 (2): 291–304. дои : 10.1016/j.cell.2006.08.039 . ПМИД 17055431 . S2CID 8123837 .
- ^ Барнс А.М., Чанг В., Морелло Р., Кабрал В.А., Вейс М., Эйр Д.Р. и др. (декабрь 2006 г.). «Дефицит хрящевого белка при рецессивном летальном несовершенном остеогенезе» . Медицинский журнал Новой Англии . 355 (26): 2757–2764. doi : 10.1056/NEJMoa063804 . ПМК 7509984 . ПМИД 17192541 .
- ^ Кабрал В.А., Чанг В., Барнс А.М., Вейс М., Скотт М.А., Лейкин С. и др. (март 2007 г.). «Дефицит пролил-3-гидроксилазы 1 вызывает рецессивное метаболическое заболевание костей, напоминающее летальный/тяжелый несовершенный остеогенез» . Природная генетика . 39 (3): 359–365. дои : 10.1038/ng1968 . ПМК 7510175 . ПМИД 17277775 .
- ^ ван Дейк Ф.С., Несбитт И.М., Цвикстра Э.Х., Никкельс П.Г., Пирсма С.Р., Фратантони С.А. и др. (октябрь 2009 г.). «Мутации PPIB вызывают тяжелый несовершенный остеогенез» . Американский журнал генетики человека . 85 (4): 521–527. дои : 10.1016/j.ajhg.2009.09.001 . ПМЦ 2756556 . ПМИД 19781681 .
- ^ Кристиансен Х.Э., Шварце У., Пайотт С.М., АлСвайд А., Аль Балви М., Альрашид С. и др. (март 2010 г.). «Гомозиготность по миссенс-мутации в SERPINH1, который кодирует белок-шаперон коллагена HSP47, приводит к тяжелому рецессивному несовершенному остеогенезу» . Американский журнал генетики человека . 86 (3): 389–398. дои : 10.1016/j.ajhg.2010.01.034 . ПМЦ 2833387 . ПМИД 20188343 .
- ^ Аланай Ю., Авайган Х., Камачо Н., Утине Г.Е., Бодуроглу К., Актас Д. и др. (апрель 2010 г.). «Мутации в гене, кодирующем белок RER FKBP65, вызывают аутосомно-рецессивный несовершенный остеогенез» . Американский журнал генетики человека . 86 (4): 551–559. дои : 10.1016/j.ajhg.2010.02.022 . ПМК 2850430 . ПМИД 20362275 .
- ^ Сэм Дж. Э., Дхармалингам М. (2017). «Несовершенный остеогенез» . Индийский журнал эндокринологии и метаболизма . 21 (6): 903–908. дои : 10.4103/ijem.IJEM_220_17 . ПМК 5729682 . ПМИД 29285457 .
- ^ Шапиро-младший (2014). «Клинико-генетическая классификация несовершенного остеогенеза и эпидемиология». Несовершенный остеогенез . Эльзевир. стр. 15–22. дои : 10.1016/b978-0-12-397165-4.00002-2 . ISBN 9780123971654 .
- ^ Jump up to: а б Сэм Дж. Э., Дхармалингам М. (1 ноября 2017 г.). «Несовершенный остеогенез» . Индийский журнал эндокринологии и метаболизма . 21 (6): 903–908. дои : 10.4103/ijem.IJEM_220_17 . ПМК 5729682 . ПМИД 29285457 .
- ^ Шахин Р., Алазами А.М., Альшаммари М.Дж., Факейх Э., Альхашми Н., Муса Н. и др. (октябрь 2012 г.). «Исследование аутосомно-рецессивного несовершенного остеогенеза в Аравии выявило новый локус, определяемый мутацией TMEM38B». Журнал медицинской генетики . 49 (10): 630–635. doi : 10.1136/jmedgenet-2012-101142 . ПМИД 23054245 . S2CID 35542661 .
- ^ Jump up to: а б Линдал К., Острём Э., Драгомир А., Симоенс С., Коук П., Ларссон С. и др. (сентябрь 2018 г.). «Гомозиготность по преждевременному стоп-кодону CREB3L1 в первом случае несовершенного рецессивного остеогенеза, связанного с недостаточностью OASIS для выживания в младенчестве» . Кость . 114 : 268–277. дои : 10.1016/j.bone.2018.06.019 . ПМИД 29936144 . S2CID 49406925 . Архивировано из оригинала 29 августа 2021 года . Проверено 29 августа 2021 г.
- ^ Келлер Р.Б., Тран Т.Т., Пайотт С.М., Пепин М.Г., Саварираян Р., МакГилливрей Г. и др. (апрель 2018 г.). «Моноаллельный и биаллельный вариант CREB3L1 вызывают легкий и тяжелый несовершенный остеогенез соответственно» . Генетика в медицине . 20 (4): 411–419. дои : 10.1038/gim.2017.115 . ПМЦ 5816725 . ПМИД 28817112 .
- ^ Мендоса-Лондоно Р., Фахиминия С., Маевски Дж., Тетро М., Надаф Дж., Канну П. и др. (июнь 2015 г.). «Рецессивный несовершенный остеогенез, вызванный миссенс-мутациями в SPARC» . Американский журнал генетики человека . 96 (6): 979–985. дои : 10.1016/j.ajhg.2015.04.021 . ПМЦ 4457955 . ПМИД 26027498 .
- ^ Дойард М., Бакро С., Хубер С., Ди Рокко М., Гольденберг А., Аглан М.С. и др. (апрель 2018 г.). « Мутации FAM46A ответственны за аутосомно-рецессивный несовершенный остеогенез» . Журнал медицинской генетики . 55 (4). БМЖ : 278–284. doi : 10.1136/jmedgenet-2017-104999 . ПМИД 29358272 . S2CID 4283476 . Архивировано из оригинала 29 августа 2021 года . Проверено 29 августа 2021 г.
- ^ Jump up to: а б Линдерт У., Кабрал В.А., Аусаварат С., Тонгкобпетч С., Людин К., Барнс А.М. и др. (июль 2016 г.). «Мутации MBTPS2 вызывают дефектно-регулируемый внутримембранный протеолиз при Х-сцепленном несовершенном остеогенезе» . Природные коммуникации . 7 (1): 11920. Бибкод : 2016NatCo...711920L . дои : 10.1038/ncomms11920 . ПМЦ 4935805 . ПМИД 27380894 .
- ^ Мозес С., Ямамото Г.Л., Гарбес Л., Кеупп К., Белеза-Мейрелеш А., Морено К.А. и др. (октябрь 2019 г.). «Аутосомно-рецессивные мутации при МЭСР вызывают несовершенный остеогенез» . Американский журнал генетики человека . 105 (4): 836–843. дои : 10.1016/j.ajhg.2019.08.008 . ПМК 6817720 . ПМИД 31564437 .
- ^ Jump up to: а б с д и Байерс П.Х., Пайотт С.М. (15 декабря 2012 г.). «Рецессивно наследуемые формы несовершенного остеогенеза» . Ежегодный обзор генетики . 46 (1): 475–497. doi : 10.1146/annurev-genet-110711-155608 . ISSN 0066-4197 . ПМИД 23145505 .
- ^ Хо Дуй Б., Житник Л., Маасалу К., Кяндла И., Пранс Е., Рейманн Е. и др. (август 2016 г.). «Мутационный анализ генов COL1A1 и COL1A2 у вьетнамских пациентов с несовершенным остеогенезом» . Геномика человека . 10 (1): 27. дои : 10.1186/s40246-016-0083-1 . ПМЦ 4983065 . ПМИД 27519266 .
- ^ Паломо Т., Виласа Т., Лазаретти-Кастро М. (декабрь 2017 г.). «Несовершенный остеогенез: диагностика и лечение». Современное мнение в эндокринологии, диабете и ожирении . 24 (6): 381–388. doi : 10.1097/MED.0000000000000367 . ПМИД 28863000 . S2CID 4555427 .
- ^ Валадарес Э.Р., Карнейру Т.Б., Сантос П.М., Оливейра А.С., Забель Б. (18 июля 2014 г.). «Что нового в генетике и несовершенстве классификации остеогенеза?» . Журнал педиатрии . 90 (6): 536–541. дои : 10.1016/j.jped.2014.05.003 . ПМИД 25046257 .
- ^ Jump up to: а б с Форлино А., Кабрал В.А., Барнс А.М., Марини Дж.К. (июнь 2011 г.). «Новые взгляды на несовершенный остеогенез» . Обзоры природы. Эндокринология . 7 (9): 540–557. дои : 10.1038/nrendo.2011.81 . ПМЦ 3443407 . ПМИД 21670757 .
- ^ Коромани Ф, Траяноска К, Риваденейра Ф, Оэй Л (4 июня 2019 г.). «Последние достижения в генетике переломов при остеопорозе» . Границы эндокринологии . 10 :337.дои : 10.3389 /fendo.2019.00337 . ПМК 6559287 . ПМИД 31231309 .
- ^ Рорбах М., Джунта С. (август 2012 г.). «Рецессивный несовершенный остеогенез: клинические, радиологические и молекулярные данные» . Американский журнал медицинской генетики. Часть C. Семинары по медицинской генетике . 160С (3): 175–189. дои : 10.1002/ajmg.c.31334 . ПМИД 22791419 . S2CID 28592112 .
- ^ Jump up to: а б Марини Дж. К., Райх А., Смит С. М. (август 2014 г.). «Несовершенный остеогенез вследствие мутаций неколлагеновых генов: уроки биологии костеобразования» . Современное мнение в педиатрии . 26 (4): 500–507. дои : 10.1097/MOP.0000000000000117 . ПМЦ 4183132 . ПМИД 25007323 .
- ^ Ханагата Н. (март 2016 г.). «Мутации IFITM5 и несовершенный остеогенез». Журнал костного и минерального обмена . 34 (2): 123–131. дои : 10.1007/s00774-015-0667-1 . ПМИД 26031935 . S2CID 3173191 .
- ^ Jump up to: а б с Пайотт С.М., Пепин М.Г., Шварце У., Ян К., Смит Дж., Байерс П.Х. (февраль 2011 г.). «Рецидив перинатального летального несовершенного остеогенеза у сибсов: анализ риска между родительским мозаицизмом для доминантных мутаций и аутосомно-рецессивным наследованием» . Генетика в медицине . 13 (2): 125–130. дои : 10.1097/GIM.0b013e318202e0f6 . ПМИД 21239989 .
- ^ Раух Ф., Глориё Ф.Х. (апрель 2004 г.). «Несовершенный остеогенез». Ланцет . 363 (9418): 1377–1385. дои : 10.1016/S0140-6736(04)16051-0 . ПМИД 15110498 . S2CID 24081895 .
- ^ Jump up to: а б с Готьери А., Узель С., Весентини С., Редаелли А., Бюлер М.Дж. (август 2009 г.). «Молекулярные и мезомасштабные механизмы несовершенного остеогенеза коллагеновых фибрилл» . Биофизический журнал . 97 (3): 857–865. Бибкод : 2009BpJ....97..857G . дои : 10.1016/j.bpj.2009.04.059 . ПМК 2718154 . ПМИД 19651044 .
- ^ Jump up to: а б с Сяо Дж., Ченг Х., Сильва Т., Баум Дж., Бродский Б. (декабрь 2011 г.). «Миссенс-мутации несовершенного остеогенеза в коллагене: структурные последствия замены глицина на аланин в сильно заряженном участке» . Биохимия . 50 (50): 10771–10780. дои : 10.1021/bi201476a . ПМК 3292618 . ПМИД 22054507 .
- ^ Jump up to: а б Нейхейс В.Х., Иствуд Д.М., Олгроув Дж., Хвид И., Вейнанс Х.Х., Банк РА, Саккерс Р.Дж. (февраль 2019 г.). «Современные концепции несовершенного остеогенеза: структура кости, биомеханика и медицинское управление» . Журнал детской ортопедии . 13 (1): 1–11. дои : 10.1302/1863-2548.13.180190 . ПМК 6376438 . ПМИД 30838070 .
- ^ Эль-Собки Т.А., Шауки Р.М., Сакр Х.М., Эльсайед С.М., Эльсайед Н.С., Рагеб С.Г., Гамаль Р. (15 ноября 2017 г.). «Систематизированный подход к рентгенологической оценке часто встречающихся генетических заболеваний костей у детей: иллюстрированный обзор» . J Musculoskelet Surg Res . 1 (2): 25. doi : 10.4103/jmsr.jmsr_28_17 . S2CID 79825711 .
- ^ Джеймсон-младший, Альберт С.И., Бусс Б., Смит П.А., Харрис Г.Ф. (29 марта 2013 г.). «3D-микронная визуализация сети кортикальных костных каналов при несовершенном остеогенезе (ОИ) человека». В Weaver JB, Molthen RC (ред.). Медицинская визуализация 2013: Биомедицинские применения в молекулярной, структурной и функциональной визуализации . Том. 8672. Международное общество оптики и фотоники. стр. 86721Л. дои : 10.1117/12.2007209 . S2CID 13876569 . Архивировано из оригинала 7 июня 2020 года . Проверено 26 августа 2020 г.
- ^ Вестгрен М., Гётерстрем К. (сентябрь 2015 г.). «Трансплантация стволовых клеток до рождения – реальный вариант лечения несовершенного остеогенеза?». Пренатальная диагностика . 35 (9): 827–832. дои : 10.1002/pd.4611 . ПМИД 25962526 . S2CID 10640427 .
- ^ Jump up to: а б с Пепин М.Г., Байерс П.Х. (декабрь 2015 г.). «Что должен знать каждый клинический генетик о тестировании на несовершенный остеогенез в случаях подозрения на жестокое обращение с детьми». Американский журнал медицинской генетики. Часть C. Семинары по медицинской генетике . 169 (4): 307–313. дои : 10.1002/ajmg.c.31459 . ПМИД 26566591 . S2CID 26045033 .
- ^ Кастанеда Р. (25 октября 2005 г.). «Родители получают 1 миллион долларов по делу о ложном насилии» . Вашингтон Пост . ISSN 0190-8286 . Проверено 4 февраля 2022 г.
- ^ Jump up to: а б «Что такое несовершенный остеогенез? Краткие факты: серия удобных для чтения публикаций для общественности» . НИАМС . Ноябрь 2014. Архивировано из оригинала 18 октября 2016 года . Проверено 15 октября 2016 г.
- ^ Суизи Т., Рив Б.Б., Харт Т.С., Флор МК, Доллар СМ, Гиллис АП, Този ЛЛ (февраль 2019 г.). «Учет точки зрения пациента в изучении редких заболеваний костей: идеи сообщества несовершенного остеогенеза» . Международный остеопороз . 30 (2): 507–511. дои : 10.1007/s00198-018-4690-7 . ПМК 6449303 . ПМИД 30191258 .
- ^ Глорье Ф.Х., Бишоп Нью-Джерси, Плоткин Х., Шабо Г., Лануэ Г., Трэверс Р. (октябрь 1998 г.). «Циклическое применение памидроната у детей с тяжелым несовершенным остеогенезом» . Медицинский журнал Новой Англии . 339 (14): 947–952. дои : 10.1056/NEJM199810013391402 . ПМИД 9753709 . S2CID 19316414 . Бесплатный полный текст
- ^ ДиМельо Л.А., Пикок М. (январь 2006 г.). «Двухлетнее клиническое исследование перорального алендроната по сравнению с внутривенным памидронатом у детей с несовершенным остеогенезом» . Журнал исследований костей и минералов . 21 (1): 132–140. дои : 10.1359/JBMR.051006 . ПМИД 16355282 . S2CID 12996685 .
- ^ Бишоп Н., Адами С., Ахмед С.Ф., Антон Дж., Арундел П., Буррен К.П. и др. (октябрь 2013 г.). «Ризедронат у детей с несовершенным остеогенезом: рандомизированное двойное слепое плацебо-контролируемое исследование». Ланцет . 382 (9902): 1424–1432. дои : 10.1016/S0140-6736(13)61091-0 . ПМИД 23927913 . S2CID 25559791 .
- ^ Уорд Л.М., Раух Ф. (октябрь 2013 г.). «Оральные бисфосфонаты при несовершенном остеогенезе у детей?». Ланцет . 382 (9902): 1388–1389. дои : 10.1016/S0140-6736(13)61531-7 . ПМИД 23927912 . S2CID 5872511 .
- ^ Jump up to: а б с Мина Б.Л., Паниграхи I, Марваха Р.К. (1 октября 2014 г.). «PO-0077 Дефицит витамина D у детей с несовершенным остеогенезом» . Архив болезней в детстве . 99 (Приложение 2): А275. дои : 10.1136/archdischild-2014-307384.747 . ISSN 0003-9888 . S2CID 72491188 . Архивировано из оригинала 18 сентября 2021 года . Проверено 18 сентября 2021 г.
- ^ Jump up to: а б Эдуард Т., Глориё Ф.Х., Раух Ф. (октябрь 2011 г.). «Предикторы и корреляты статуса витамина D у детей и подростков с несовершенным остеогенезом» . Журнал клинической эндокринологии и метаболизма . 96 (10): 3193–3198. дои : 10.1210/jc.2011-1480 . ПМИД 21832107 .
- ^ Самбрано МБ, Бризола Э, Пиньейру Б, Ванц А.П., Мелло ЭД, Феликс ТМ (18 мая 2016 г.). «Изучение детерминант статуса витамина D у педиатрических пациентов с несовершенным остеогенезом». Журнал Американского колледжа питания . 35 (4): 339–345. дои : 10.1080/07315724.2015.1057776 . ПМИД 26709914 . S2CID 45930763 .
- ^ Ротшильд Л., Голлер Дж.К., Воронов П., Барабанова А., Смит П. (ноябрь 2018 г.). «Анестезия у детей с несовершенным остеогенезом: ретроспективный обзор карт 83 пациентов и 205 анестетиков за 7 лет» . Детская анестезия . 28 (11): 1050–1058. дои : 10.1111/pan.13504 . ПМИД 30295359 . S2CID 52929810 . Архивировано из оригинала 29 августа 2021 года . Проверено 29 августа 2021 г.
- ^ Хоук Дж.П. (2014). «Несовершенный остеогенез» . В Хоук Дж.П., Хаче М., Сан Л.С. (ред.). Справочник по детской анестезии . Нью-Йорк: Образование Макгроу-Хилл . ISBN 9780071769358 . Архивировано из оригинала 29 августа 2021 года . Проверено 29 августа 2021 г.
- ^ Голлер Дж.К., Ротшильд Л. (2020), Крузе Р.В. (редактор), «Анестезирующее и послеоперационное обезболивание» (ePub) , Несовершенный остеогенез: Руководство по принятию хирургических решений и уходу на основе конкретных случаев , Springer International Publishing, стр. . 111–125, номер домена : 10.1007/978-3-030-42527-2_8 , ISBN. 978-3-030-42527-2 , S2CID 219515776
- ^ Jump up to: а б Бехера П., Сантоши Дж.А., Гиваругезе Н.М., Мина Великобритания, Селванаягам Р. (28 февраля 2020 г.). «Смещение телескопического гвоздя из эпифиза: описание случая с анализом вероятного механизма» . Куреус . 12 (2): е7130. дои : 10.7759/cureus.7130 . ISSN 2168-8184 . ПМК 7105006 . ПМИД 32257675 .
- ^ «Финансовая помощь» . Детская больница Шрайнерс . Архивировано из оригинала 3 марта 2021 года . Проверено 21 августа 2021 г.
Детская больница Шрайнерс стремится предоставлять специализированную помощь детям с ортопедическими [...] заболеваниями независимо от платежеспособности семей.
- ^ «Лидер в лечении несовершенного остеогенеза (НО)» . Шрайнерс Интернэшнл . Архивировано из оригинала 28 сентября 2007 года . Проверено 5 июля 2007 г.
- ^ Софилд Х.А., Миллар Э.А. (1 декабря 1959 г.). «Фрагментация, перестройка и интрамедуллярная стержневая фиксация деформаций длинных костей у детей: десятилетняя оценка» . Журнал костной и суставной хирургии . 41 (8): 1371–1391. дои : 10.2106/00004623-195941080-00001 . ISSN 0021-9355 .
- ^ «Телескопическая система IM Фасье-Дюваля» . Пега Медикал . Лаваль, Квебек . Архивировано из оригинала 15 августа 2021 года . Проверено 15 августа 2021 г.
- ^ Jump up to: а б Стериан А., Баланеску Р., Барбилиан А., Уличи А. (2015). «Остеосинтез при несовершенном остеогенезе, телескопические и нетелескопические гвозди» . Журнал медицины и жизни . 8 (4): 563–565. ПМК 4656972 . ПМИД 26664490 .
- ^ Jump up to: а б Фрасье Ф., Дюваль П., Пейли Д. (2021). «Хирургическая техника телескопической системы ИМ Фасье-Дюваля» (PDF) . Пега Медикал . Лаваль, Квебек . FD-ST-EN Редакция K. Архивировано (PDF) из оригинала 15 августа 2021 года . Проверено 15 августа 2021 г.
- ^ Беля К.М., Нокс Дж.Б. (1 января 2020 г.). «Спондилодез у детей с несовершенным остеогенезом: общенациональное ретроспективное сравнительное когортное исследование за 12-летний период» . Современная ортопедическая практика . 31 (1): 72–75. дои : 10.1097/BCO.0000000000000805 . ISSN 1940-7041 . S2CID 209229412 .
- ^ Зани А., Форд-Адамс М., Рэтклифф М., Беван Д., Инге Т.Х., Десаи А. (январь 2017 г.). «Операция по снижению веса улучшает качество жизни педиатрических пациентов с несовершенным остеогенезом» . Хирургия ожирения и связанных с ним заболеваний . 13 (1). Elsevier Inc .: 41–44. дои : 10.1016/j.soard.2015.11.029 . ПМЦ 5965274 . ПМИД 26948942 .
- ^ Семлер О, Фрике О, Везироглу К, Старк С, Шенау Э (1 января 2007 г.). «Предварительные результаты подвижности после вибрации всего тела у обездвиженных детей и подростков» . Журнал скелетно-мышечных и нейрональных взаимодействий . 7 (1): 77–81. ПМИД 17396011 . Архивировано из оригинала 18 августа 2021 года . Проверено 18 августа 2021 г.
- ^ Jump up to: а б Бирия М., Аббас Ф.М., Мозаффар С., Ахмади Р. (июль 2012 г.). «Несовершенный дентиногенез, связанный с несовершенным остеогенезом» . Журнал стоматологических исследований . 9 (4): 489–494. ПМЦ 3491340 . ПМИД 23162594 .
- ^ Jump up to: а б с д Плит Дж. (18 декабря 2020 г.). «Переработка отходов окупается для Mereo» . Оцените Vantage . Архивировано из оригинала 18 сентября 2021 года . Проверено 18 сентября 2021 г.
- ^ Дельгадо-Калле Дж., Сато А.Ю., Беллидо Т. (март 2017 г.). «Роль и механизм действия склеростина в кости» . Кость . 96 : 29–37. дои : 10.1016/j.bone.2016.10.007 . ПМЦ 5328835 . ПМИД 27742498 .
- ^ Глориё Ф.Х., Девогелаер Дж.П., Дуригова М., Гоэмер С., Хемсли С., Якоб Ф. и др. (июль 2017 г.). «Антитело к склеростину BPS804 у взрослых с умеренным несовершенным остеогенезом: результаты рандомизированного исследования фазы 2а» . Журнал исследований костей и минералов . 32 (7): 1496–1504. дои : 10.1002/jbmr.3143 . ПМИД 28370407 . S2CID 4432023 .
- ^ «Ultragenyx и Mereo BioPharma объявляют о сотрудничестве и лицензионном соглашении по использованию сетрусумаба при несовершенном остеогенезе» . Ultragenyx Pharmaceutical Inc. 17 декабря 2020 г. Архивировано из оригинала 18 сентября 2021 г. Проверено 18 сентября 2021 г.
- ^ «Исследование на взрослых пациентах с несовершенным остеогенезом I, III или IV типа, получавших лечение BPS804 (ASTEROID)» . ClinicalTrials.gov . Национальная медицинская библиотека США . 1 апреля 2021 г. NCT03118570 . Проверено 18 сентября 2021 г.
- ^ Купер М., Грегорек Л. (25 ноября 2019 г.). «Сетрусумаб готов приступить к решающим исследованиям» . Троицкая Дельта . Архивировано из оригинала 18 сентября 2021 года . Проверено 18 сентября 2021 г.
- ^ Харди А. (24 сентября 2020 г.). «Мерео сетрусумаб получил статус редкого педиатрического заболевания FDA как несовершенный остеогенез» . Биовторники . Архивировано из оригинала 18 сентября 2021 года . Проверено 18 сентября 2021 г.
- ^ «Исследование по оценке ромосозумаба у детей и подростков с несовершенным остеогенезом» . ClinicalTrials.gov . Национальная медицинская библиотека США. 23 июля 2021 г. NCT04545554. Архивировано из оригинала 18 сентября 2021 года . Проверено 18 сентября 2021 г.
- ^ По неведению (2 сентября 2022 г.). «Акции Ultragenyx: нисходящая траектория, вероятно, продолжится» . В поисках Альфа . Проверено 24 сентября 2022 г.
- ^ Карпентер Т., Гулански Б., Баррос Дж.С. (21 апреля 2023 г.). «Исследование по оценке эффективности и безопасности сетрусумаба у участников с несовершенным остеогенезом» . Йельская медицина . Архивировано из оригинала 26 июля 2023 года . Проверено 16 декабря 2023 г.
- ^ Грин Р (11 марта 2012 г.). «Хрупкая болезнь костей заставила мать сделать «нелегкий выбор» » . Хартфорд Курант . Проверено 16 сентября 2021 г.
- ^ Хоппер Т (6 февраля 2013 г.). « «Несовместимо с жизнью»: рассказ матери о ее трудном решении попросить об аборте на позднем сроке проливает свет на дебаты о живорождении» . Национальная почта . Проверено 16 сентября 2021 г.
- ^ Патерсон Ч.Р., Огстон С.А., Генри Р.М. (февраль 1996 г.). «Продолжительность жизни при несовершенном остеогенезе» . БМЖ . 312 (7027): 351. doi : 10.1136/bmj.312.7027.351 . ПМК 2350292 . ПМИД 8611834 .
- ^ Плоткин Х. (29 февраля 2016 г.). «Генетика несовершенного остеогенеза» . Архивировано из оригинала 30 декабря 2010 года.
- ^ «2.4: Частота заболеваний костей» . Здоровье костей и остеопороз: отчет главного хирурга (PDF) . Роквилл, Мэриленд : Управление главного хирурга США , Министерство здравоохранения и социальных служб США . 2004. с. 68. МЫ 225 B71259 2004.
- ^ «Панель несовершенного остеогенеза» . Университета Небраски Медицинский центр . Архивировано из оригинала 30 июля 2019 года . Проверено 30 июля 2019 г.
- ^ Абали С., Арман А., Атай З., Берекет А., Бас С., Халилоглу Б. и др. (19 августа 2016 г.). «Частота рецессивного несовершенного остеогенеза в турецкой когорте и генетические причины» . Рефераты ESPE . 86 . Bioscientifica: 131. Архивировано из оригинала 26 августа 2021 года . Проверено 26 августа 2021 г.
- ^ Ловенштейн Э.Дж. (май 2009 г.). «Несовершенный остеогенез у 3000-летней мумии» . Нервная система ребенка . 25 (5): 515–516. дои : 10.1007/s00381-009-0817-7 . ПМИД 19212769 .
- ^ Jump up to: а б с Тейнмонт Дж (2007). «История несовершенного остеогенеза или болезни хрупкой кости: несколько остановок на пути длиной в 3000 лет» . Б-Энт . 3 (3): 157–173. ПМИД 17970442 . Архивировано из оригинала 29 августа 2021 года . Проверено 29 августа 2021 г.
- ^ Груневельд Э (12 ноября 2018 г.). «Ивар Бескостный» . Энциклопедия всемирной истории . Ограниченная энциклопедия древней истории. Архивировано из оригинала 21 августа 2021 года . Проверено 21 августа 2021 г.
- ^ Льюис Р., Лота Г., Гаур А. (8 января 2019 г.). «Ивар Бескостный» . Британская энциклопедия . Архивировано из оригинала 24 июня 2021 года . Проверено 21 августа 2021 г.
- ^ Мальбранш Н. (1688 г.). (на французском языке) (изд. 1842 г.). п. 119 – через Wikisource .
- ^ Эделу Б., Нду И., Асиноби И., Обу Х., Адимора Г. (март 2014 г.). «Несовершенный остеогенез: описание случая и обзор литературы» . Анналы исследований в области медицины и здравоохранения . 4 (Приложение 1). Нигерийская медицинская ассоциация : S1–S5. дои : 10.4103/2141-9248.131683 . ПМК 4083720 . ПМИД 25031897 .
- ^ Гектоен Л. (май 1903 г.). «Анатомическое исследование карлика с короткими конечностями с особым упором на несовершенный остеогенез и хондродистрофию плода» . Американский журнал медицинских наук . 125 (5). Дж. Б. Липпинкотт и компания : 751–769. дои : 10.1097/00000441-190305000-00001 . ISBN 978-0-243-38392-4 . S2CID 71836609 . Архивировано из оригинала 16 августа 2021 года . Проверено 16 августа 2021 г.
- ^ Адэр-Дайтон, Калифорния (1912). «Четыре поколения голубых склеротиков» . Офтальмоскоп . 10 . Лондон: Джордж Пулман и сыновья, Ltd: 188–189. OCLC 1049881577 – через Интернет-архив .
- ^ Вролик В (1849). Tabulae ad illusanderam hominis et mlememium, tam naturalem quam abnormem — Плоды людей и млекопитающих, изображенные и описанные в их правильном и нерегулярном развитии [ Иллюстрации эмбриогенеза человека и млекопитающих embreogenesin , как нормального, так и аномального ] (на латыни и нидерландском языке. Амстердам: GMP Лондонк. стр. 562–566. OCLC 489920691 . Архивировано из оригинала 29 августа 2021 года . Проверено 29 августа 2021 г.
- ^ Jump up to: а б Балджет Б (январь 2002 г.). «Аспекты истории несовершенного остеогенеза (синдрома Вролика)» . Анналы анатомии — Anatomischer Anzeiger . 184 (1): 1–7. дои : 10.1016/S0940-9602(02)80023-1 . ПМИД 11876477 . Архивировано из оригинала 12 июня 2018 года . Проверено 29 августа 2021 г.
- ^ Балджет Б (2002). «Виллем Вролик и «его» синдром» . В: Раушманн М.А., Томанн К.Д., Зихнер Л. (ред.). История рубежей ортопедии . Том 4. Гейдельберг: Verlag Theodor Steinkopff . стр. 133–144. дои : 10.1007/978-3-642-57510-5_15 . ISBN 978-3-642-57510-5 . Архивировано из оригинала 8 июня 2018 года . Проверено 29 августа 2021 г.
- ^ Лузер Е (1906). остеогенеза (так называемого «идиопатического остеопсатироза») « О осознании несовершенного врожденного и позднего . Послания с рубежа медицины и хирургии (на немецком языке). 15 : 161-207. Архивировано из оригинала 21 августа 2021 года . Проверено 21 августа 2021 г.
- ^ Synd/1743 в Who Named It?
- ^ Расмуссен С., Дэнкс А. (2010). Двойная спираль, двойная радость: Дэвид Дэнкс, отец клинической генетики в Австралии . Карлтон, Виктория : Издательство Мельбурнского университета. п. 116. ИСБН 978-0-522-85799-3 . OCLC 906592657 . Архивировано из оригинала 28 августа 2021 года . Проверено 28 августа 2021 г.
- ^ Jump up to: а б Варман М.Л., Кормье-Дэр В., Холл С., Краков Д., Лахман Р., ЛеМеррер М. и др. (май 2011 г.). «Нозология и классификация генетических заболеваний скелета: редакция 2010 г.» . Американский журнал медицинской генетики. Часть А. 155А (5): 943–968. дои : 10.1002/ajmg.a.33909 . ПМК 3166781 . ПМИД 21438135 .
- ^ Jump up to: а б «Несовершенный остеогенез (НО) у такс» . Лаборатория ветеринарной генетики Калифорнийского университета в Дэвисе . Школа ветеринарной медицины Калифорнийского университета в Дэвисе . Архивировано из оригинала 28 августа 2021 года . Проверено 28 августа 2021 г.
- ^ Jump up to: а б «Несовершенный остеогенез (О.И. Даксунд)» . Центр генетики животных . Дженератио ГмбХ. Архивировано из оригинала 8 ноября 2018 года . Проверено 7 ноября 2018 г.
- ^ Jump up to: а б с д Эндерли Т.А., Берч С.Р., Темплет Дж.Н., Карриеро А. (сентябрь 2016 г.). «Животные модели несовершенного остеогенеза: применение в клинических исследованиях» . Ортопедические исследования и обзоры . 8 : 41–55. дои : 10.2147/ORR.S85198 . ПМК 6209373 . ПМИД 30774469 .
- ^ Сабан Дж., Зуссман М.А., Хэви Р., Патвардхан А.Г., Шнайдер ГБ, Кинг Д. (декабрь 1996 г.). «Гетерозиготные мыши OIM демонстрируют легкую форму несовершенного остеогенеза» . Кость . 19 (6): 575–579. дои : 10.1016/S8756-3282(96)00305-5 . ПМИД 8968022 . Архивировано из оригинала 30 июня 2018 года . Проверено 28 августа 2021 г.
Внешние ссылки
[ редактировать ]- «Обзор несовершенного остеогенеза» . NIH Остеопороз и связанные с ним заболевания костей — Национальный ресурсный центр . Национальные институты здравоохранения, Министерство здравоохранения и социальных служб США. 8 мая 2023 г.
| Базы данных органов управления : Национальные |
|---|