Медуллярная кистозная болезнь почек
| Медуллярная кистозная болезнь почек | |
|---|---|
 | |
| Медуллярная кистозная болезнь почек имеет аутосомно-доминантный тип наследования. | |
| Специальность | Медицинская генетика |
| Симптомы | Полидипсия [ 1 ] |
| Типы | МККД1 и МККД2 [ 2 ] [ 3 ] |
| Метод диагностики | Биопсия почки, УЗИ почек, общий анализ крови [ 4 ] |
| Медикамент | На данный момент лечения нет, Пейте много жидкости, Соляные добавки [ 4 ] |
Медуллярная кистозная болезнь почек ( МКБП ) — аутосомно-доминантное заболевание почек, характеризующееся тубулоинтерстициальным склерозом, приводящее к терминальной стадии заболевания почек . Поскольку наличие кист не является ни ранним, ни типичным диагностическим признаком заболевания, и поскольку по крайней мере четыре различные генные мутации могут привести к этому состоянию, название аутосомно-доминантное тубулоинтерстициальное заболевание почек ( ADTKD было предложено добавить ). с основным генетическим вариантом для конкретного человека. [ 5 ] [ 6 ] Важно отметить, что если кисты обнаруживаются в собирательных трубочках мозгового вещества, они могут привести к уменьшению почки, в отличие от поликистозной болезни почек . [ нужна ссылка ] Существуют две известные формы медуллярной кистозной болезни почек: муцин-1 болезнь почек 1 (MKD1) и муцин-2 болезнь почек/уромодулиновая болезнь почек (MKD2). [ 1 ] Третья форма заболевания возникает из-за мутаций в гене, кодирующем ренин (ADTKD-REN), и ранее была известна как семейная ювенильная гиперурикемическая нефропатия 2-го типа. [ 7 ]
Признаки и симптомы
[ редактировать ]Что касается признаков/симптомов медуллярной кистозной болезни почек, это заболевание нелегко диагностировать и оно встречается редко. В этом состоянии потеря функции почек происходит медленно с течением времени, однако у больного могут наблюдаться следующие признаки/симптомы: [ 1 ]
- Полидипсия
- Энурез
- Слабость
- Отсутствие аппетита
- Зуд
- Боль в костях
- Бледность
- Тошнота
У некоторых людей с этим заболеванием развивается подагра . [ 8 ] Это состояние, при котором у пациентов возникают сильная боль и отек большого пальца ноги или другого сустава, например колена. При отсутствии лечения заболевание становится хроническим и поражает суставы большую часть времени, а не периодически. [ 9 ]
Причина
[ редактировать ]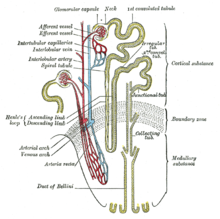
Нормальные люди имеют две копии генов MUC1 и UMOD . Гены продуцируют белок муцин-1 и уромодулин соответственно. Эти белки экспрессируются только в определенных клетках почки – толстом восходящем колене Генле и дистальных извитых канальцах – обеих частях почечных канальцев. Белок покрывает поверхность канальцев и защищает канальцы. [ 2 ] [ 3 ] [ 1 ] При MKD у людей имеется один нормальный и один аномальный MUC1 ген . При уромодулиновой болезни почек (UKD) у людей имеется один нормальный и один аномальный UMOD ген . Аномальный (мутировавший) ген продуцирует неправильно свернутый белковый продукт, который откладывается в клетках почечных канальцев (в части клетки, называемой эндоплазматической сетью). Аномальный белок накапливается в клетке и вызывает ее медленную гибель, что приводит к окончательной потере функции почек, что проявляется в виде болезненного состояния. [ 10 ] [ 11 ]
Заболевание почек MKD1/муцин-1
[ редактировать ]
Заболевание почек муцина-1 (МКД) возникает из-за мутации гена MUC1 , который расположен на хромосоме 1 . [ 12 ] Белок муцин-1 участвует в создании слизеподобного вещества, которое покрывает поверхность различных мелких канальцев в организме. Он экспрессируется на клетках дистальных канальцев в почках. Заболевание почек муцина-1 (МКД) вызвано мутацией гена MUC1 . [ 12 ]
Эта мутация изменяет генетическую последовательность гена MUC1 , что приводит к появлению нового мутированного белка. Более того, это заболевание встречается крайне редко, и его распространенность неизвестна. Это заболевание является аутосомно-доминантным , что означает, что оно характеризуется 50% вероятностью наследования и медленно прогрессирующим хроническим заболеванием почек, что приводит к необходимости диализа или трансплантации почки . Люди с мутацией в этом гене могут иметь различную степень потери функции почек: некоторые люди начинают проходить диализ в возрасте тридцати лет, тогда как другие могут не идти на диализ до более позднего возраста. [ 10 ] [ 13 ]
Ранее это заболевание называлось медуллярной кистозной болезнью почек 1-го типа. Оно было названо так потому, что у некоторых людей с этим заболеванием были кисты (небольшие отверстия) в средней части (мозговом веществе) почек. С тех пор было обнаружено, что эти кисты встречаются редко и не обнаруживаются у большинства пациентов с мутациями MUC1. По этой причине от этого названия отказались, ученые (входящие в группу «Инициативы по диализу почек» и «Глобальные результаты»), специализирующиеся на этом заболевании, формально объединились и создали официальное название для этого и подобных состояний. [ 6 ]
Это состояние было обозначено как аутосомно-доминантное тубулоинтерстициальное заболевание почек (АДТБП). При аутосомно-доминантном наследовании только один родитель должен быть затронут, чтобы передать заболевание. Тубулоинтерстициальный используется потому, что проблемы, возникающие при этом заболевании, являются следствием атрофии канальцев петли Генле и фиброза медуллярного интерстиция . [ нужна медицинская ссылка ]
MKD2/уромодулин заболевание почек
[ редактировать ]
Медуллярная кистозная болезнь почек 2 типа возникает из-за мутаций гена UMOD на , хромосоме 16 который кодирует белок уромодулин. [ 11 ] [ 14 ] Это заболевание также является аутосомно-доминантным , что означает, что оно характеризуется 50% вероятностью наследования и медленно прогрессирующим хроническим заболеванием почек, что приводит к необходимости диализа или трансплантации почки . У людей с этим заболеванием подагра возникает относительно рано в жизни. Мутации в этом гене могут привести к различной степени потери функции почек. [ 8 ]
При наследовании MKD2, как и MKD1, аутосомно-доминантный тип наследования указывает на то, что 50% детей больного человека унаследуют заболевание. «Тубулоинтерстициальный» используется потому, что проблемы, возникающие при этом заболевании, происходят в отделе почки, известном как тубулоинтерстиций. Большинство людей используют более короткое название заболевания — уромодулиновая болезнь почек (УКД). [ 15 ]
Диагностика
[ редактировать ]
Диагноз медуллярной кистозной болезни почек можно поставить посредством физикального осмотра. [ 4 ] Дальнейшие тесты/обследования заключаются в следующем: [ 1 ]
- обычный анализ крови, называемый сывороточным креатинином Можно сделать . Креатинин является продуктом распада мышц, поскольку функция почек снижается, количество креатинина в крови увеличивается. Таким образом, у большинства пострадавших людей симптомы MCKD отсутствуют, но они обнаруживают, что это заболевание возникает из-за повышения уровня креатинина в крови. [ 16 ]
- У больных также наблюдается повышение уровня мочевой кислоты в крови . При MCKD почкам трудно избавиться от мочевой кислоты. О том, что уровень мочевой кислоты в крови повышен, можно узнать, сдав анализ крови. Подагра вызвана высоким уровнем мочевой кислоты, поэтому пациенты часто страдают подагрой. [ 17 ]
- почек УЗИ в этом состоянии обычно показывает нормальные или небольшие по размеру почки (иногда присутствуют кисты). Однако, поскольку кисты присутствуют у многих нормальных людей, эти кисты не помогают в постановке диагноза, поэтому можно провести биопсию почки , чтобы определить, есть ли у человека это заболевание. Биопсия почки — это процедура, при которой игла вводится в почку и удаляет небольшой кусочек почечной ткани. Затем эту ткань исследуют под микроскопом. [ 18 ] [ 19 ]
- Окончательное тестирование и диагноз MCKD можно провести путем анализа мутаций гена UMOD, это можно сделать с помощью анализа крови. [ 20 ]
Уход
[ редактировать ]Что касается лечения медуллярной кистозной болезни почек, в настоящее время не существует специфических методов лечения этого заболевания, а также не существует известных диет, замедляющих прогрессирование заболевания. Однако лечение симптомов можно решить следующим образом: эритропоэтин используется для лечения анемии , а гормон роста используется, когда рост становится проблемой. Кроме того, в какой-то момент может потребоваться пересадка почки. Наконец, необходимо сократить употребление продуктов, содержащих калий и фосфат. [ 1 ]
Исследовать
[ редактировать ]Ученые из Института Броуда (Кембридж, Массачусетс) определили генетическую причину UKD как мутации в гене MUC1 . [ 12 ]
См. также
[ редактировать ]- Нефронофтиз – другое генетическое заболевание почек, имеющее сходные патологические и клинические проявления, но наследственное по аутосомно-рецессивному типу, часто с более ранним началом и может проявлять характерные внепочечные проявления. [ 21 ]
Ссылки
[ редактировать ]- ^ Перейти обратно: а б с д и ж Девараджан, Прасад. Лэнгман, Крейг Б. (ред.). «Лечение и ведение медуллярных кистозных заболеваний: медицинская помощь, хирургическая помощь, консультации» . Электронная медицина . МедСкейп. Архивировано из оригинала 23 октября 2020 года . Проверено 13 августа 2024 г.
- ^ Перейти обратно: а б «Запись OMIM — № 174000 — МЕДУЛЛЯРНАЯ КИСТИЗНАЯ БОЛЕЗНЬ ПОЧЕК 1; MCKD1» . omim.org . Архивировано из оригинала 12 декабря 2019 года . Проверено 2 января 2018 г.
- ^ Перейти обратно: а б «Запись OMIM — № 603860 — МЕДУЛЛЯРНАЯ КИСТИЧНАЯ БОЛЕЗНЬ ПОЧЕК 2; MCKD2» . omim.org . Архивировано из оригинала 10 марта 2017 года . Проверено 2 января 2018 г.
- ^ Перейти обратно: а б с «Медуллярная кистозная болезнь почек: Медицинская энциклопедия MedlinePlus» . www.nlm.nih.gov . Архивировано из оригинала 19 мая 2024 г. Проверено 3 июня 2016 г.
- ^ Торрес, Винсент Э.; Харрис, Питер С. (2016). «Кистозные заболевания почек». В Скорецки, Карл; Чертоу, Гленн М.; Марсден, Филип А; Таал, Мартен В.; Ю, Алан С.Л. (ред.). «Почка» Бреннера и ректора (10-е изд.). Филадельфия, Пенсильвания: Эльзевир. п. 1504. ИСБН 978-1-4557-4836-5 .
Поскольку развитие медуллярных кист не является ни ранним, ни типичным признаком заболевания, многие теперь считают термин «медуллярная кистозная болезнь почек» неправильным и предложили термин «аутосомно-доминантная тубулоинтерстициальная болезнь почек» (АДТБП) для обозначения группы заболевания, вызванные как минимум четырьмя генами: MUC1, кодирующим мукопротеин муцин-1 (MUC-1) (хромосомная локализация 1q22); UMOD, кодирующий уромодулин, также известный как белок Тамма-Хорсфолла (хромосома 16p12.3); HNF1B, кодирующий ядерный фактор гепатоцитов-1β (хромосома 17q12); и REN, кодирующий ренин (хромосома 1q32.1).
- ^ Перейти обратно: а б Экардт, КУ; Альпер, СЛ; Антиньяк, К; Блейер, Эй Джей; Шово, Д; Дахан, К; Дельты, С; Хоскинг, А; Кмох, С; Рампольди, Л; Визенер, М; Вольф, Монтана; Девюйст, О (октябрь 2015 г.). «Аутосомно-доминантное тубулоинтерстициальное заболевание почек: диагностика, классификация и лечение — консенсусный отчет KDIGO» . Почки Интернешнл . 88 (4): 676–83. дои : 10.1038/ki.2015.28 . ПМИД 25738250 . – через ScienceDirect (может потребоваться подписка или контент может быть доступен в библиотеках.)
- ^ Кмоч, Станислав; Живна, Мартина; Блейер, Энтони (1993). «Аутосомно-доминантное тубулоинтерстициальное заболевание почек, связанное с REN (ADTKD-REN)» . Джин Обзоры . Вашингтонский университет, Сиэтл. Архивировано из оригинала 28 октября 2020 года . Проверено 11 июня 2016 г.
- ^ Перейти обратно: а б Блейер, AJ; Вудард, AS.; Шихаби, З.; Сандху, Дж.; Чжу, Х.; Сатко, С.Г.; Веллер, Н.; Детердинг, Э.; Макбрайд, Д. (июнь 2003 г.). «Клиническая характеристика семьи с мутацией гена уромодулина (гликопротеина Тамм-Хорсфолла)» . Почки Int . 64 (1): 36–42. дои : 10.1046/j.1523-1755.2003.00081.x . ПМИД 12787393 .
- ^ «Подагра: МедлайнПлюс» . www.nlm.nih.gov . Архивировано из оригинала 5 июля 2016 г. Проверено 15 июня 2016 г.
- ^ Перейти обратно: а б Справочник, Дом генетики. «Медуллярная кистозная болезнь почек 1 типа» . Домашний справочник по генетике . Архивировано из оригинала 30 сентября 2020 г. Проверено 15 июня 2016 г.
- ^ Перейти обратно: а б Справочник, Дом генетики. «уромодулин-ассоциированное заболевание почек» . Домашний справочник по генетике . Архивировано из оригинала 22 сентября 2020 г. Проверено 15 июня 2016 г.
- ^ Перейти обратно: а б с Кирби, А. (март 2013 г.). «Мутации, вызывающие медуллярную кистозную болезнь почек типа 1, лежат в большом VNTR в MUC1, пропущенном при массовом параллельном секвенировании» . Природная генетика . 45 (3): 299–303. дои : 10.1038/ng.2543 . ПМК 3901305 . ПМИД 23396133 .
- ^ Блейер, Энтони Дж.; Кмох, Станислав (1 января 1993 г.). «Аутосомно-доминантное тубулоинтерстициальное заболевание почек, связанное с MUC1 (ADTKD-MUC1)» . В Пагоне, Роберта А.; Адам, Маргарет П.; Ардингер, Холли Х.; Уоллес, Стефани Э.; Амемия, Энн; Бин, Лора Дж. Х.; Берд, Томас Д.; Фонг, Чин-То; Меффорд, Хизер С. (ред.). Джин Обзоры . Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл. ПМИД 23946964 . Архивировано из оригинала 13 августа 2020 г. Проверено 4 сентября 2017 г. обновление 2013
- ^ Харт, TC; Горри, MC; Харт, PS; Вудард, А.С.; Шихаби, З.; Сандху, Дж.; Рубашки, Б.; Сюй, Л.; Чжу, Х. (1 декабря 2002 г.). «Мутации гена UMOD ответственны за медуллярную кистозную болезнь почек 2 и семейную ювенильную гиперурикемическую нефропатию» . Журнал медицинской генетики . 39 (12): 882–892. дои : 10.1136/jmg.39.12.882 . ISSN 1468-6244 . ПМК 1757206 . ПМИД 12471200 .
- ^ Блейер, Энтони Дж.; Харт, П. Сюзанна (1 января 1993 г.). «Аутосомно-доминантное тубулоинтерстициальное заболевание почек, связанное с UMOD (ADTKD-UMOD)» . В Пагоне, Роберта А.; Адам, Маргарет П.; Ардингер, Холли Х.; Уоллес, Стефани Э.; Амемия, Энн; Бин, Лора Дж. Х.; Берд, Томас Д.; Фонг, Чин-То; Меффорд, Хизер С. (ред.). Джин Обзоры . Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл. ПМИД 20301530 . Архивировано из оригинала 2 мая 2020 г. Проверено 4 сентября 2017 г. обновление 2013
- ^ «Анализ крови на креатинин: Медицинская энциклопедия MedlinePlus» . www.nlm.nih.gov . Архивировано из оригинала 19 мая 2024 г. Проверено 15 июня 2016 г.
- ^ «Мочевая кислота – кровь: Медицинская энциклопедия MedlinePlus» . www.nlm.nih.gov . Архивировано из оригинала 5 июля 2016 г. Проверено 15 июня 2016 г.
- ^ «Биопсия почки» . www.niddk.nih.gov . Архивировано из оригинала 17 июня 2016 г. Проверено 15 июня 2016 г.
- ^ «УЗИ брюшной полости: Медицинская энциклопедия MedlinePlus» . www.nlm.nih.gov . Архивировано из оригинала 28 мая 2016 г. Проверено 15 июня 2016 г.
- ^ Справочник, Дом генетики. «УМОД» . Домашний справочник по генетике . Архивировано из оригинала 9 августа 2016 г. Проверено 15 июня 2016 г.
- ^ Девараджан, Прасад (16 марта 2020 г.). Лэнгман, Крейг Б. (ред.). «Медуллярная кистозная болезнь: обзор» . Медскейп . Проверено 13 августа 2024 г.
Дальнейшее чтение
[ редактировать ]- Хатебур, Ник (2001). «Подтверждение локуса гена медуллярной кистозной болезни почек (MCKD2) на хромосоме 16p12» . Почки Интернешнл . 60 (4): 1233–1239. дои : 10.1046/j.1523-1755.2001.00932.x . ПМИД 11576337 .
- Хильдебрандт, Фридхельм ; Отто, Эдгар (1 сентября 2000 г.). «Молекулярная генетика нефронофтиза и медуллярной кистозной болезни почек» . Журнал Американского общества нефрологов . 11 (9): 1753–1761. дои : 10.1681/ASN.V1191753 . ISSN 1046-6673 . ПМИД 10966501 . Проверено 15 июня 2016 г.