Прогрессирующая оссифицирующая фибродисплазия
Эта статья нуждается в дополнительных цитатах для проверки . ( сентябрь 2023 г. ) |
| Прогрессирующая оссифицирующая фибродисплазия | |
|---|---|
| Другие имена | Болезнь каменного человека, болезнь Мюнхмейера. |
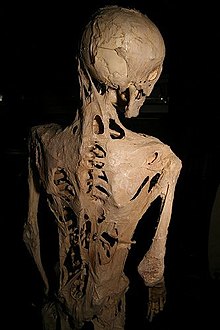 | |
| Скелет Гарри Рэймонда Истлака , пациента с прогрессирующей оссифицирующей фибродисплазией, с характерным аномальным ростом костей. | |
| Специальность | Медицинская генетика , ревматология |
| Симптомы | Непрерывный рост костей |
| Обычное начало | До 10 лет |
| Дифференциальный диагноз | Фиброзная дисплазия |
| Уход | Никто |
| Прогноз | Средняя продолжительность жизни составляет около 40 лет (при правильном управлении). |
| Частота | 801 подтвержденный случай во всем мире (2017 г.); уровень заболеваемости оценивается в 0,5 случаев на миллион человек (1 на 2 миллиона) |
фибродисплазия прогрессива ( / ˌ f aɪ b roʊ ; d ɪ ˈ s p l eɪ ʒ ( i ) ə ɒ ˈ s ɪ f ɪ k æ n z p r ə ˈ ɡ r ɛ s ɪ v ə / Оссифицирующая [1] сокр. ФОП ), также называемый болезнью Мюнхмейера или ранее прогрессирующий оссифицирующий миозит , представляет собой чрезвычайно редкое заболевание соединительной ткани , при котором волокнистая соединительная ткань, такая как мышцы , сухожилия и связки, превращается в костную ткань . Это единственное известное заболевание, при котором одна система органов переходит в другую. [2] Это тяжелое инвалидизирующее расстройство, от которого невозможно избавиться.
ФОП вызван мутацией гена ACVR1 . Мутация влияет на механизм восстановления организма, вызывая фиброзной ткани, мышцы , сухожилия и связки , окостенение включая либо спонтанно, либо при повреждении в результате травмы. Во многих случаях незначительные травмы могут привести суставов к необратимому сращению в виде новой кости, заменяющей поврежденную мышечную ткань. Это новое костное образование (известное как «гетеротопическая оссификация») в конечном итоге образует вторичный скелет и постепенно ограничивает способность пациента двигаться. Кость, образовавшаяся в результате этого процесса, идентична «нормальной» кости, просто находится в неправильном месте. Косвенные данные свидетельствуют о том, что это заболевание может вызывать деградацию суставов отдельно от характерного для них роста костей. [3]
Было показано, что хирургическое удаление лишней кости приводит к тому, что организм «восстанавливает» пораженный участок дополнительной костью. Хотя скорость роста костей может различаться в зависимости от пациента, в конечном итоге это заболевание приводит к обездвиживанию больных, поскольку новая кость заменяет мускулатуру и срастается с существующим скелетом. За это ФОП получил прозвище « болезнь каменного человека ». [4]
Признаки и симптомы
[ редактировать ]По неизвестным причинам дети, рожденные с ФОП, часто имеют деформированные большие пальцы ног , иногда отсутствует сустав или, в других случаях, просто наблюдается заметное уплотнение на малом суставе. [5] Первое «обострение», приводящее к образованию кости ФОП, обычно происходит в возрасте до 10 лет. [5] Рост костей обычно происходит от верхней части тела вниз, так же, как кости растут у плода. У ребенка с ФОП обычно развиваются дополнительные кости, начиная с шеи, затем в плечах, руках, области груди и, наконец, в ступнях. [6]
В частности, оссификация обычно сначала наблюдается в дорсальной, аксиальной, краниальной и проксимальной областях тела. В дальнейшем заболевание прогрессирует в вентральном, аппендикулярном, каудальном и дистальном отделах. [5] Однако это не обязательно происходит в указанном порядке из-за обострений, вызванных травмой. Нередко опухолевидные образования, характеризующие обострение заболевания, появляются внезапно.
Рост костей, происходящий во время обострений, может привести к потере подвижности пораженных суставов, в том числе, если вовлекается челюсть/нижняя челюсть, к неспособности полностью открыть рот, ограничению речи и приема пищи. Конкретное обострение этого состояния в суставах стопы/голеностопного сустава может привести к ограничению возможности поставить ногу на землю. [ нужна ссылка ] Рост костей также может привести к иммобилизации бедра или колена, что влияет на способность человека ходить. Дополнительное образование костей вокруг грудной клетки ограничивает расширение легких и диафрагмы, вызывая респираторные осложнения. [5]
Поскольку это заболевание встречается невероятно редко и встречается только у 1 из 2 миллионов человек, его можно ошибочно диагностировать как рак или фиброз . Это может привести к тому, что врачи назначат биопсию , что потенциально усугубит рост кости ФОП. [7] Наличие деформированных пальцев ног или больших пальцев у детей, рожденных с ФОП, помогает отличить это заболевание от других проблем со скелетом. [8]
При правильном медицинском лечении средний возраст выживания составляет 40 лет. Однако поздняя диагностика, травмы и инфекции могут сократить продолжительность жизни. [9]
Причины
[ редактировать ]ФОП вызывается аутосомно- доминантным аллелем на хромосоме 2q23-24. [10] Аллель имеет переменную экспрессивность , но полную пенетрантность . Большинство случаев вызвано спонтанной мутацией гамет ; большинство людей с ФОП не могут или предпочитают не иметь детей. Похожее, но менее катастрофическое заболевание — фиброзная дисплазия , вызванная постзиготической мутацией .
За заболевание ответственна мутация в гене ACVR1 (также известном как активинподобная киназа 2 (ALK2)). [10] ACVR1 кодирует рецептор активина типа 1, рецептор BMP типа 1. Мутация вызывает замену кодона 206 с аргинина на гистидин в белке ACVR1. [11] [12] Эта замена вызывает аномальную активацию ACVR1, приводящую к трансформации соединительной и мышечной ткани во вторичный скелет. Это заставляет эндотелиальные клетки трансформироваться в мезенхимальные стволовые клетки , а затем в кость. [13] рецептора активина типа 1 В норме ген ACVR1 кодирует трансмембранную киназу , которая связывает рецепторы BMP (BMPR типа I и BMPR типа II) для передачи сигналов хондрогенеза . BMP принадлежат к суперсемейству белков, известных как белки трансформирующего фактора роста-бета ( TGF- β ). Связывание белка ACVR1 с рецепторами BMP запускает сигнальный каскад, который имеет решающее значение для индукции формирования эндохондральной кости во время развития, а также гомеостаза скелета и тканей . [14]
Генетика
[ редактировать ]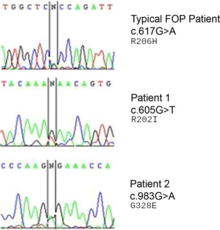
ФОП является аутосомно-доминантным заболеванием. Таким образом, вероятность заболевания у ребенка больного гетерозиготного родителя и незатронутого родителя составляет 50%. Два больных человека могут родить здоровых детей. Два здоровых человека могут произвести на свет пораженное потомство в результате мутации гена. Гомозиготная доминантная форма протекает более тяжело, чем гетерозиготная форма. [15]
Белок, вызывающий оссификацию, обычно деактивируется ингибирующим белком после того, как кости плода формируются в утробе матери, но у пациентов с ФОП белок продолжает работать. Аберрантное формирование кости у пациентов с ФОП происходит, когда поврежденная соединительная ткань или мышечные клетки в местах повреждения или роста неправильно экспрессируют фермент для восстановления кости во время апоптоза (саморегулируемая гибель клеток), в результате чего лимфоциты содержат избыток костного морфогенетического белка 4 (BMP4). ) обеспечивается во время реакции иммунной системы. Возникающая в результате кость возникает независимо от нормального скелета, образуя свои собственные дискретные элементы скелета. Однако эти элементы могут сливаться с нормальной скелетной костью. [16] При этом процессе затрагиваются диафрагма, язык и внеглазные мышцы, а также сердечная и гладкая мускулатура . [5] Поскольку неправильный фермент остается нерешенным в рамках иммунного ответа, организм продолжает поставлять неправильные BMP4-содержащие лимфоциты. BMP4 – это продукт, который способствует развитию скелета нормального эмбриона. [17]
Ген ACVR1 кодирует рецептор костного морфогенного белка (BMP); этот ген мутирован в ФОП. Он отвечает за рост и развитие костей и мышц. Типичная мутация R202H заставляет ингибитор FKBP1A менее прочно связываться с петлей активации GS. [18] В результате ACVR1 не отключается эффективно, происходит разрастание костей и хрящей и сращение суставов. [19] Атипичные мутации, затрагивающие другие остатки, действуют аналогичным образом. В некоторых случаях рецептор может сигнализировать о своей активности, не будучи связанным со своим активирующим лигандом. [20]
Большинство случаев ФОП были результатом мутации нового гена: у этих людей не было истории этого конкретного расстройства в их семье. В некоторых случаях человек унаследовал мутацию от одного пораженного родителя. [19]
Диагностика
[ редактировать ]Как правило, ФОП можно диагностировать с помощью рентгенограмм . Ранняя диагностика этого заболевания с помощью радиологии очень важна, чтобы избежать ненужных инвазивных исследований, таких как биопсия . Самая маленькая или незначительная травма или внутримышечные инъекции могут усилить прогрессирование заболевания через воспаление, отсюда и преимущество радиологии. Клиницисты должны знать об этом редком заболевании, поскольку его часто ошибочно принимают за рак или другие доброкачественные образования, такие как инфекция, что приводит к биопсии, которая часто может ускорить прогрессирование заболевания. [21]
Вспышки можно измерить клинически по повышенным уровням щелочной фосфатазы и костно-специфической щелочной фосфатазы . [22] Другим характерным признаком ФОП является укороченный большой палец стопы с деформацией дистальной части первой плюсневой кости и отсутствием или аномалией первой фаланги и/или межфалангового сустава. [23]
Уход
[ редактировать ]Хотя ФОП остается неизлечимым, многообещающий прорыв заключается в одобренном лечении Сохонос ( паловаротен ). [24] Примечательно, что попытки хирургического удаления кости у пациента с ФОП могут привести к взрывному росту новой кости. [25] Во время проведения анестезии люди с ФОП могут столкнуться с трудностями при интубации , рестриктивной болезнью легких , изменениями в электропроводящей системе сердца . [26] Следует избегать действий, повышающих риск падения или повреждения мягких тканей, поскольку даже незначительная травма может спровоцировать гетеротопическое окостенение . [27]
Хотя не существует эффективных окончательных методов лечения этого расстройства, существуют периодические методы лечения, такие как противовоспалительные препараты для подавления воспаления в результате обострений или воспаления из-за повреждения мышц. В настоящее время хирургическое вмешательство обычно не рекомендуется людям с ФОП, поскольку оно может спровоцировать быстрое костеобразование в местах разрезов или там, где на мышечные или соединительные ткани наложены швы. Можно рассмотреть возможность хирургического вмешательства, спасающего жизнь, однако передовой практикой может считаться разработка плана хирургического вмешательства с участием специалиста по ФОП. Хирургическое устранение контрактур суставов обычно оказывается безуспешным и сопряжено с риском новой гетеротопической оссификации, вызванной травмой. [28]
Эпидемиология
[ редактировать ]По состоянию на 2017 год [update]Во всем мире подтверждено около 800 случаев ФОП, что делает ФОП одним из самых редких известных заболеваний. [29] По оценкам, заболеваемость ФОП составляет 0,5 случая на миллион человек и затрагивает все этнические группы. [29]
История
[ редактировать ]

Медицинские отчеты, описывающие людей, страдающих ФОП, относятся к доктору Гаю Патину в 1692 году. [29] Первоначально ФОП назывался прогрессирующим оссифицирующим миозитом и считалось, что он вызван мышечным воспалением ( миозитом ), которое вызывает образование кости. [29] Заболевание было переименовано Виктором А. МакКьюсиком мягкие ткани, помимо мышц (например, связки ). в 1970 году после открытия того, что этому заболеванию также поражаются и [29]
Самый известный случай ФОП — дело Гарри Истлака (1933–1973). Его состояние начало развиваться в возрасте десяти лет, и к моменту его смерти от пневмонии в ноябре 1973 года, за шесть дней до 40-летия, его тело полностью окостенело, и он мог двигать только губами. Истлак никогда при жизни не встречал другого человека с ФОП. [30]
Истлак пожертвовал свое тело науке, а его скелет сейчас находится в Музее Мюттера в Филадельфии и оказался бесценным источником информации в изучении ФОП. Другой человек с ФОП, Кэрол Орзел (20 апреля 1959 г. - февраль 2018 г.), также пожертвовала свое тело музею, и ее скелет был выставлен там, рядом с скелетом Истлака, в феврале 2019 года. [31] [32]
Исследовать
[ редактировать ]

Клинические испытания изотретиноина , этидроната с пероральными кортикостероидами и малеата пергексилина не продемонстрировали эффективности, хотя вариабельное течение заболевания и небольшая распространенность вызывают неопределенность. [22]
Несколько фармацевтических компаний, занимающихся редкими заболеваниями, в настоящее время находятся на разных стадиях исследования различных терапевтических подходов к ФОП.
США В августе 2015 года Управление по разработке орфанных продуктов Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) предоставило La Jolla Pharmaceuticals статус орфанного препарата для двух новых соединений для лечения ФОП. Эти соединения представляют собой низкомолекулярные ингибиторы протеинкиназы, предназначенные для избирательного блокирования ACVR1 (ALK2). [38]
В августе 2015 года компания Clementia Pharmaceuticals также начала набор детей (в возрасте 6 лет и старше) в клиническое исследование фазы II по изучению паловаротена для лечения ФОП. [39] Доклинические исследования показали, что паловаротен, гамма-агонист рецептора ретиноевой кислоты, блокирует аномальное образование костей на животных моделях путем ингибирования вторичных информационных систем в пути BMP. [40] Клементия лицензировала паловаротен у компании Roche Pharmaceuticals, которая ранее оценивала это соединение у более чем 800 человек, включая здоровых добровольцев и пациентов с хронической обструктивной болезнью легких. Паловаротен получил статус ускоренного режима от FDA и статус орфанного препарата для лечения ФОП как от FDA, так и от Европейского агентства по лекарственным средствам (EMA). [39]
В сентябре 2015 года компания Regeneron объявила о новом понимании механизма заболевания, включающем активацию рецептора ACVR1 активином А. В 2016 году компания начала исследование первой фазы своего антитела к активину, REGN 2477 , на здоровых добровольцах; исследование фазы 2 на пациентах с ФОП было проведено в 2017 году. [41]
Другой потенциальный терапевтический подход включает аллель-специфическую РНК-интерференцию , которая нацелена на деградацию мутировавшей мРНК, сохраняя при этом нормальную экспрессию гена ACVR1. [42]
Дальнейшие исследования механизмов гетеротопического формирования кости при ФОП могут помочь в разработке методов лечения других нарушений, связанных с формированием внескелетной кости.
Фибро/адипогенные предшественники (FAP) могут быть типом болезнетворных клеток, ответственным за активин А-зависимое эктопическое образование кости как в мышцах, так и в сухожилиях мышей, несущих FOP, вызывающий мутацию ACVR1 (R206H). [43]
В декабре 2019 года Ipsen частично приостановила клиническое лечение для людей в возрасте до 14 лет из-за сообщений о раннем сращении пластинок роста.
В последнее время, по состоянию на 2021 г. [update]Потенциальный терапевтический кандидат, саракатиниб , находится на стадии III клинических испытаний в качестве мощного ингибитора гетеротопической оссификации на мышах дикого типа и мутантных мышах ACVR1. [44]
См. также
[ редактировать ]- Международная ассоциация ФОП
- Несовершенный остеогенез - группа генетических нарушений, приводящих к хрупкости костей, состоянию, характеризующемуся необычно легкой ломкостью костей, вызванному мутациями в генах, отвечающих за ФОП.
- Прогрессирующая костная гетероплазия – редкое генетическое заболевание, характеризующееся кожным или подкожным оссификацией.
- FOP Friends - Страницы британской благотворительной организации,
Ссылки
[ редактировать ]- ^ «Медицинское определение прогрессирующей оссифицирующей фибродисплазии» . www.merriam-webster.com . Проверено 8 мая 2022 г.
- ^ Каплан, Фредерик С.; Шен, Ци; Лунев Виталий; Зееманн, Петра; Гроппе, Джей; Катагири, Такенобу; Пиньоло, Роберт Дж.; Шор, Эйлин М. (2008). «Скелетные метаморфозы при прогрессирующей оссифицирующей фибродисплазии (ФОП)» . Журнал костного и минерального метаболизма . 26 (6): 521–530. дои : 10.1007/s00774-008-0879-8 . ПМК 3620015 . ПМИД 18979151 .
- ^ Пинковски, Джен (1 марта 2019 г.). «Вот что происходит, когда ткани вашего тела превращаются в кости» . Нэшнл Географик . Архивировано из оригинала 3 марта 2019 года.
- ^ Верма, Амит Кумар; Ага, Паллави; Сингх, Шайлеш Кумар; Сингх, Рагини (2012). «Болезнь каменного человека: прогрессирующая оссифицирующая фибродисплазия: новый взгляд на визуализацию» . Отчеты о случаях BMJ . 2012 : bcr2012006422. дои : 10.1136/bcr-2012-006422 . ПМЦ 4543882 . ПМИД 22843760 .
- ^ Jump up to: Перейти обратно: а б с д и Каплан, Фредерик С.; Ле Меррер, Мартина; Глейзер, Дэвид Л.; Пиньоло, Роберт Дж.; Голдсби, Роберт Э.; Киттерман, Джозеф А.; Гроппе, Джей; Шор, Эйлин М. (март 2008 г.). «Прогрессирующая оссифицирующая фибродисплазия» . Передовая практика и исследования в области клинической ревматологии . 22 (1): 191–205. дои : 10.1016/j.berh.2007.11.007 . ПМК 2424023 . ПМИД 18328989 .
- ^ «Прогрессирующая оссифицирующая фибродисплазия: MedlinePlus Genetics» . medlineplus.gov . Проверено 31 декабря 2022 г.
- ^ Обамуйиде, штат Ха; Огунладе, СО (март 2015 г.). «Опухоль, для которой хирургическое вмешательство принесет больше вреда, чем пользы: отчет о случае прогрессирующей оссифицирующей фибродисплазии» . Нигерийский медицинский журнал для последипломного образования . 22 (1): 83–8. дои : 10.4103/1117-1936.163373 . ПМИД 25875418 . S2CID 33239264 . ПроКвест 1850178336 .
- ^ «Прогрессирующая оссифицирующая фибродисплазия» . Национальный центр биомедицинских коммуникаций Листера Хилла . Проверено 12 декабря 2013 г.
- ^ Салех, Мохаммед; Командор Йост; Бокарди, Рената; Кинабо, Грейс; Хамель, Бен (2015). «Прогрессирующая оссифицирующая фибродисплазия с незначительной односторонней аномалией большого пальца стопы в спорадическом случае из Северной Танзании с распространенной мутацией ACVR1c.617G>A» . Панафриканский медицинский журнал . 22 : 299. дои : 10.11604/pamj.2015.22.299.8032 . ПМЦ 4769042 . ПМИД 26966495 .
- ^ Jump up to: Перейти обратно: а б Шор, Эйлин М; Сюй, Мэйци; Фельдман, Джордж Дж; Фенстермахер, Дэвид А; Чо, Тэ Джун; Чой, Ин Хо; Коннор, Дж. Майкл; Делай, Патрисия; Глейзер, Дэвид Л; ЛеМеррер, Мартина; Морхарт, Рольф; Роджерс, Джон Дж; Смит, Роджер; Триффит, Джеймс Т; Уртисбереа, Дж. Андони; Заслов, Майкл; Браун, Мэтью А; Каплан, Фредерик С. (май 2006 г.). «Рекуррентная мутация рецептора BMP I типа ACVR1 вызывает наследственную и спорадическую прогрессирующую оссифицирующую фибродисплазию». Природная генетика . 38 (5): 525–527. дои : 10.1038/ng1783 . ПМИД 16642017 . S2CID 41579747 .
- ^ «Исследователи Пенсильванского университета обнаружили ген, создающий второй скелет» (пресс-релиз). Медицинский факультет Пенсильванского университета. 23 апреля 2006 г.
- ^ Хатселл, Сара Дж.; Готово, Винсент; Волкен, Дана М. Алесси; Хуанг, Лили; Ким, Хён Дж.; Ван, Лили; Вэнь, Сялин; Наннуру, Калян С.; Хименес, Джоанна; Се, Лицинь; Дас, Нандита; Махул, Женевьева; Черноморский, Ростислав; Д'Амброзио, Дэвид; Корпина, Ричард А.; Шенхерр, Кристофер Дж.; Фили, Киран; Ю, Пол Б.; Янкопулос, Джордж Д.; Мерфи, Эндрю Дж.; Экономидес, Арис Н. (2 сентября 2015 г.). «Мутация рецептора ACVR1 R206H вызывает прогрессирующую оссифицирующую фибродисплазию, придавая чувствительность к активину А» . Наука трансляционной медицины . 7 (303): 303ра137. doi : 10.1126/scitranslmed.aac4358 . ПМК 6164166 . ПМИД 26333933 .
- ^ ван Динтер, Мартен; Виссер, Нильс; де Гортер, Дэвид Джей-Джей; Доорн, Джойс; Гуманс, Мари-Жозе; де Бур, Ян; тен Дейке, Питер (23 ноября 2009 г.). «Мутация ALK2 R206H, связанная с прогрессивной оссифицирующей фибродисплазией, придает конститутивную активность рецептору BMP типа I и повышает чувствительность мезенхимальных клеток к индуцированной BMP дифференцировке остеобластов и формированию кости» . Журнал исследований костей и минералов . 25 (6): 091211115834058–35. дои : 10.1359/jbmr.091110 . ПМИД 19929436 . S2CID 207269687 .
- ^ Линь, Шусянь; Свобода, Кэти К.Х.; Фэн, Цзянь Ц.; Цзян, Синьцюань (5 апреля 2016 г.). «Биологическая функция рецепторов I типа костного морфогенетического белка в кости» . Исследование костей . 4 (1): 16005. doi : 10.1038/boneres.2016.5 . ISSN 2095-6231 . ПМЦ 4820739 . ПМИД 27088043 .
- ^ Каммингс, Майкл Р. Человеческая наследственность: принципы и проблемы Cengage Learning, 2011, [2009]. п. 77
- ^ Шор, Эйлин М.; Каплан, Фредерик С. (2008). «Итоги редкого генетического нарушения формирования внескелетной кости, прогрессирующей оссифицирующей фибродисплазии (ФОП)» . Кость . 43 (3): 427–443. дои : 10.1016/j.bone.2008.05.013 . ПМК 2601573 . ПМИД 18590993 .
- ^ Кирзенбаум, Авраам (2002). Гистология и клеточная биология . Нью-Йорк: Мосби. ISBN 978-0-323-01639-1 .
- ^ "AVCR1" , Домашний справочник по генетике , Национальная медицинская библиотека США, август 2007 г. По состоянию на 18 февраля 2014 г.
- ^ Jump up to: Перейти обратно: а б «Прогрессирующая оссифицирующая фибродисплазия» , Домашний справочник генетики , Национальная медицинская библиотека США, август 2007 г. По состоянию на 18 февраля 2014 г.
- ^ Петри, Калифорния; Ли, Вашингтон; Буллок, АН; Пойнтон, Джей-Джей; Смит, Р; Рассел, Р.Г.; Браун, Массачусетс; Вордсворт, BP; Триффит, Дж. Т. (2009). «Новые мутации в ACVR1 приводят к атипичным проявлениям у двух пациентов с прогрессирующей оссифицирующей фибродисплазией» . ПЛОС ОДИН . 4 (3): е5005. Бибкод : 2009PLoSO...4.5005P . дои : 10.1371/journal.pone.0005005 . ПМЦ 2658887 . ПМИД 19330033 .
- ^ Ямин, Гиам; Дагиги, Шади; Мафи, Махмуд (31 мая 2021 г.). «Прогрессирующая оссифицирующая фибродисплазия (ФОП) проявляется в виде быстро растущего некальцинированного образования шеи» . Журнал радиологических отчетов о случаях заболевания . 15 (5): 10–16. дои : 10.3941/jrcr.v15i5.4103 . ISSN 1943-0922 . ПМЦ 8253151 . ПМИД 34276874 .
- ^ Jump up to: Перейти обратно: а б Кито, Хироши; Ачива, Масатака; Канеко, Хироши; Мисима, Кеничи; Мацусита, Масаки; Кадоно, Изуми; Горовиц, Джон Д.; Саллюстио, Бенедетта С; Оно, Кинджи; Исигуро, Наоки (2013). «Малеат пергексилина в лечении прогрессирующей оссифицирующей фибродисплазии: открытое клиническое исследование» . Сиротский журнал редких заболеваний . 8 (1): 163. дои : 10.1186/1750-1172-8-163 . ПМК 4015865 . ПМИД 24131551 .
- ^ «Прогрессивная оссифицирующая фибродисплазия» . Август 2020.
- ^ Исследования, Центр оценки лекарств и (17 августа 2023 г.). «FDA одобрило первое лечение прогрессирующей оссифицирующей фибродисплазии» . FDA .
- ^ Американская академия хирургов-ортопедов (май 2006 г.). «Прогрессирующая оссифицирующая фибродисплазия (ФОП)» . orthinfo.aaos.org. Архивировано из оригинала 21 июня 2012 года . Проверено 7 октября 2011 г.
- ^ Ньютон, MC; Аллен, PW; Райан, округ Колумбия (февраль 1990 г.). «Прогрессивная оссифицирующая фибродисплазия» . Британский журнал анестезии . 64 (2): 246–250. дои : 10.1093/бья/64.2.246 . ПМИД 2317429 .
- ^ «Медицинское лечение прогрессирующей оссифицирующей фибродисплазии: текущие соображения по лечению» (PDF) . Международная ассоциация прогрессирующей оссифицирующей фибродисплазии. Январь 2020.
- ^ Каплан, Фредерик С.; Ле Меррер, Мартина; Глейзер, Дэвид Л.; Пиньоло, Роберт Дж.; Голдсби, Роберт; Киттерман, Джозеф А.; Гроппе, Джей; Шор, Эйлин М. (март 2008 г.). «Прогрессирующая оссифицирующая фибродисплазия» . Лучшие практики и исследования. Клиническая ревматология . 22 (1): 191–205. дои : 10.1016/j.berh.2007.11.007 . ISSN 1521-6942 . ПМК 2424023 . ПМИД 18328989 .
- ^ Jump up to: Перейти обратно: а б с д и ж г Мартелли, Андерсон; Сантос, Арнальдо Родригес (3 июля 2014 г.). «Клеточные и морфологические аспекты прогрессирующей оссифицирующей фибродисплазии: уроки формирования, восстановления и биоинженерии кости» . Органогенез . 10 (3): 303–311. дои : 10.4161/org.29206 . ПМЦ 4750545 . ПМИД 25482313 .
- ^ Каплан, Ф.С. (2013). Скелет в шкафу. Джин, 528 (1), 7–11. https://doi.org/10.1016/j.gene.2013.06.022
- ^ Маккалоу, Мари (28 февраля 2019 г.). «Новая выставка Музея Муттера исполняет последнее желание женщины, превратившейся в кость» . Филадельфийский исследователь . Проверено 28 февраля 2019 г.
- ^ «Музей Мюттера представляет новый экспонат: скелет женщины из Филадельфии с редким заболеванием костей» . Музей Мюттера . 5 марта 2019 года. Архивировано из оригинала 28 августа 2021 года . Проверено 7 марта 2020 г.
- ^ «Доктор Фредерик Каплан, доктор медицинских наук, лауреат премии Rare Impact Award 2017» . Национальная организация редких заболеваний . 15 марта 2017 года . Проверено 8 мая 2022 г.
- ^ «Фредерик Каплан: Премия за редкое воздействие» (PDF) . Альманах . 63 (32). Пенсильванский университет : 7. 25 апреля 2017 г. Проверено 8 мая 2022 г.
- ^ «Хронология ФОП» . Пенн Медисин . Пенсильванский университет . Проверено 8 мая 2022 г.
- ^ «АП: Ученые разгадали тайну редкого заболевания костей» . Новости Эн-Би-Си . 24 апреля 2006 года . Проверено 8 мая 2022 г.
- ^ Медер, Томас (1 февраля 1998 г.). «Несколько сотен человек превратились в кости» . Атлантика . Проверено 8 мая 2022 г.
- ^ «Фармацевтическая компания La Jolla получила статус орфанного препарата для двух новых соединений для лечения прогрессирующей оссифицирующей фибродисплазии» (пресс-релиз). Фармацевтическая компания Ла Хойя. 18 августа 2015 г.
- ^ Jump up to: Перейти обратно: а б «Clementia Pharmaceuticals расширяет текущее исследование фазы 2, включив в него детей с прогрессирующей оссифицирующей фибродисплазией (FOP)» (пресс-релиз). Клементия Фармасьютикалс. 25 августа 2015 г.
- ^ «Трубопровод» . www.clementiapharma.com . Архивировано из оригинала 29 сентября 2015 года . Проверено 22 ноября 2015 г.
- ^ «Regeneron делится новостями о своей текущей программе исследований ФОП» . Международная ассоциация прогрессирующей оссифицирующей фибродисплазии. 9 марта 2017 г.
- ^ Дж. В. Лоури; В. Розен (2012). «Аллель-специфическая РНК-интерференция в ФОП, подавляющая ген ФОП» . Генная терапия . 19 (7): 701–702. дои : 10.1038/gt.2011.190 . ПМИД 22130446 . S2CID 24990800 .
- ^ Лис-Шепард, Джон Б.; Ямамото, Масакадзу; Бисвас, Арпита А.; Стессель, Шон Дж.; Николас, Сара-Энн Э.; Когсвелл, Кэти А.; Девараконда, Парвати М.; Шнайдер, Майкл Дж.; Камминс, Саманта М.; Лежандр, Николас П.; Ямамото, Сёко; Каартинен, Веса; Хантер, Джеффри В.; Голдхамер, Дэвид Дж. (декабрь 2018 г.). «Активин-зависимая передача сигналов в фиброзных/адипогенных предшественниках вызывает прогрессирующую оссифицирующую фибродисплазию» . Природные коммуникации . 9 (1): 471. Бибкод : 2018NatCo...9..471L . дои : 10.1038/s41467-018-02872-2 . ПМК 5797136 . ПМИД 29396429 .
- ^ Уильямс, Элеонора; Багарова Яна; Керр, Джорджина; Ся, Донг-Донг; Плейс, Элси С.; Дей, Девавина; Шен, Юэ; Бокобо, Джеффри А.; Мохедас, Агустин Х.; Хуан, Сюли; Сандерсон, Филип Э. (2021). «Саракатиниб является эффективным клиническим кандидатом для лечения прогрессирующей оссифицирующей фибродисплазии» . JCI-инсайт . 6 (8): e95042. doi : 10.1172/jci.insight.95042 . ISSN 2379-3708 . ПМК 8119212 . ПМИД 33705358 .
Дальнейшее чтение
[ редактировать ]- Коэн, М. Майкл; Хауэлл, Робин Э. (октябрь 1999 г.). «Этиология фиброзной дисплазии и синдрома МакКьюна-Олбрайта». Международный журнал челюстно-лицевой хирургии . 28 (5): 366–371. дои : 10.1016/s0901-5027(99)80085-x . ПМИД 10535539 .
| Базы данных органов управления : Национальные |
|---|