Машинное обучение в биоинформатике
| Часть серии о |
| Искусственный интеллект |
|---|
Машинное обучение в биоинформатике — это применение алгоритмов машинного обучения в биоинформатике . [1] включая геномику , протеомику , микроматрицы , системную биологию , эволюцию и интеллектуальный анализ текста . [2] [3]
До появления машинного обучения алгоритмы биоинформатики приходилось программировать вручную; для таких задач, как предсказание структуры белка , это оказалось трудным. [4] Методы машинного обучения, такие как глубокое обучение, могут изучать особенности наборов данных, вместо того, чтобы требовать от программиста определять их индивидуально. Алгоритм может дополнительно научиться объединять низкоуровневые функции в более абстрактные функции и так далее. Этот многоуровневый подход позволяет таким системам делать сложные прогнозы при соответствующем обучении. Эти методы контрастируют с другими подходами вычислительной биологии , которые, хотя и используют существующие наборы данных, не позволяют интерпретировать и анализировать данные непредвиденным образом.
Задачи [ править ]
Алгоритмы машинного обучения в биоинформатике можно использовать для прогнозирования, классификации и выбора признаков. Методы достижения этой задачи разнообразны и охватывают многие дисциплины; наиболее известными среди них являются машинное обучение и статистика. Задачи классификации и прогнозирования направлены на построение моделей, которые описывают и различают классы или концепции для будущего прогнозирования. Различия между ними заключаются в следующем:
- Классификация/распознавание выводит категориальный класс, а прогнозирование — числовой признак.
- Тип алгоритма или процесса, используемого для построения прогнозных моделей на основе данных с использованием аналогий, правил, нейронных сетей, вероятностей и/или статистики.
Благодаря экспоненциальному росту информационных технологий и применимых моделей, включая искусственный интеллект и интеллектуальный анализ данных, в дополнение к доступу к все более полным наборам данных были созданы новые и более совершенные методы анализа информации, основанные на их способности к обучению. Такие модели позволяют выйти за рамки описания и предоставляют информацию в форме тестируемых моделей.
машинного Подходы обучения
Искусственные нейронные сети [ править ]
Искусственные нейронные сети в биоинформатике используются для: [5]
- Сравнение и сопоставление последовательностей РНК, белков и ДНК.
- Идентификация промоторов и поиск генов по последовательностям, связанным с ДНК.
- Интерпретация данных экспрессии генов и микрочипов.
- Выявление сети (регуляторной) генов.
- Изучение эволюционных связей путем построения филогенетических деревьев .
- Классификация и предсказание структуры белков .
- Молекулярный дизайн и стыковка .
Разработка функций [ править ]
Способ извлечения признаков (часто векторов в многомерном пространстве) из данных предметной области является важным компонентом систем обучения. [6] В геномике типичным представлением последовательности является вектор частот k-меров , который представляет собой вектор размерности чьи записи учитывают появление каждой подпоследовательности длины в заданной последовательности. Поскольку для такого маленького значения, как размерность этих векторов огромна (например, в этом случае размерность равна ), такие методы, как анализ главных компонентов, используются для проецирования данных в пространство более низкой размерности, таким образом выбирая меньший набор функций из последовательностей. [6] [ необходимы дополнительные ссылки ]




Классификация [ править ]
В задачах машинного обучения этого типа выходные данные представляют собой дискретную переменную. Одним из примеров задач такого типа в биоинформатике является маркировка новых геномных данных (таких как геномы некультивируемых бактерий) на основе модели уже маркированных данных. [6]
марковские Скрытые модели
Скрытые модели Маркова (HMM) — это класс статистических моделей для последовательных данных (часто связанных с системами, развивающимися с течением времени). HMM состоит из двух математических объектов: наблюдаемого процесса, зависящего от состояния. и ненаблюдаемый (скрытый) процесс состояния . В HMM процесс состояния не наблюдается напрямую – это «скрытая» (или «латентная») переменная – но наблюдения производятся за процессом, зависящим от состояния (или процессом наблюдения), который управляется основным процессом состояния ( и которое, таким образом, можно рассматривать как зашумленное измерение интересующих состояний системы). [7] СММ могут быть сформулированы в непрерывном времени. [8] [9]


HMM можно использовать для профилирования и преобразования множественного выравнивания последовательностей в систему оценки по конкретной позиции, подходящую для удаленного поиска в базах данных гомологичных последовательностей. [10] Кроме того, экологические явления могут быть описаны с помощью HMM. [11]
Сверточные нейронные сети [ править ]
Сверточные нейронные сети (CNN) — это класс глубоких нейронных сетей , архитектура которых основана на общих весах ядер или фильтров свертки, которые скользят вдоль входных признаков, обеспечивая трансляционно-эквивариантные ответы, известные как карты признаков. [12] [13] CNN используют преимущества иерархической структуры данных и собирают шаблоны возрастающей сложности, используя более мелкие и простые шаблоны, обнаруженные с помощью их фильтров. [14]
Сверточные сети были вдохновлены биологическими . процессами [15] [16] [17] [18] в том, что структура связей между нейронами напоминает организацию зрительной коры животных . Отдельные кортикальные нейроны реагируют на стимулы только в ограниченной области поля зрения, известной как рецептивное поле . Рецептивные поля разных нейронов частично перекрываются и охватывают все поле зрения.
CNN использует относительно небольшую предварительную обработку по сравнению с другими алгоритмами классификации изображений . Это означает, что сеть учится оптимизировать фильтры (или ядра) посредством автоматического обучения, тогда как в традиционных алгоритмах эти фильтры разрабатываются вручную . Эта меньшая зависимость от предварительных знаний аналитика и вмешательства человека при ручном извлечении признаков делает CNN желательной моделью. [14]
Филогенетическая сверточная нейронная сеть (Ph-CNN) представляет собой архитектуру сверточной нейронной сети, предложенную Фиоранти и др. в 2018 году для классификации данных метагеномики . [19] В этом подходе филогенетические данные наделяются святоотеческим расстоянием (сумма длин всех ветвей, соединяющих две операционные таксономические единицы [OTU]) для выбора k-окрестностей для каждой OTU, а каждая OTU и ее соседи обрабатываются сверточными фильтрами.
Самостоятельное обучение [ править ]
В отличие от контролируемых методов, методы самоконтролируемого обучения изучают представления, не полагаясь на аннотированные данные. Это хорошо подходит для геномики, где методы высокопроизводительного секвенирования могут создавать потенциально большие объемы немаркированных данных. Некоторые примеры методов самостоятельного обучения, применяемых в геномике, включают DNABERT и Self-GenomeNet. [20] [21]
Случайный лес [ править ]
Случайные леса (RF) классифицируются путем построения ансамбля деревьев решений и вывода среднего прогноза для отдельных деревьев. [22] Это модификация бутстрап-агрегирования (которая объединяет большую коллекцию деревьев решений) и может использоваться для классификации или регрессии . [23] [24]
Поскольку случайные леса дают внутреннюю оценку ошибки обобщения, перекрестная проверка не требуется. Кроме того, они создают близости, которые можно использовать для вменения отсутствующих значений и которые позволяют по-новому визуализировать данные. [25]
С вычислительной точки зрения случайные леса привлекательны, поскольку они естественным образом поддерживают как регрессию, так и (многоклассовую) классификацию, относительно быстро обучаются и прогнозируют, зависят только от одного или двух параметров настройки, имеют встроенную оценку ошибки обобщения, могут использоваться. непосредственно для задач большой размерности и может быть легко реализован параллельно. По статистике, случайные леса привлекательны дополнительными функциями, такими как меры переменной важности, дифференциальное взвешивание классов, вменение пропущенных значений, визуализация, обнаружение выбросов и обучение без учителя. [25]
Кластеризация [ править ]
Кластеризация — разделение набора данных на непересекающиеся подмножества так, чтобы данные в каждом подмножестве были как можно ближе друг к другу и как можно дальше от данных в любом другом подмножестве в соответствии с некоторой определенной расстояния или сходства функцией . общий метод статистического анализа данных.
Кластеризация занимает центральное место во многих исследованиях в области биоинформатики, основанных на данных, и служит мощным вычислительным методом, с помощью которого средства иерархической, центроидной, основанной на распределении, основанной на плотности и самоорганизующейся классификации карт уже давно изучаются и используются в классических машинах. настройки обучения. В частности, кластеризация помогает анализировать неструктурированные и многомерные данные в виде последовательностей, выражений, текстов, изображений и т. д. Кластеризация также используется для получения информации о биологических процессах на геномном уровне, например, о функциях генов, клеточных процессах, подтипах клеток, регуляции генов и метаболических процессах. [26]
используемые в биоинформатике кластеризации , Алгоритмы
Алгоритмы кластеризации данных могут быть иерархическими или секционными. Иерархические алгоритмы находят последовательные кластеры, используя ранее созданные кластеры, тогда как алгоритмы разделения определяют все кластеры сразу. Иерархические алгоритмы могут быть агломеративными (снизу вверх) или разделительными (сверху вниз).
Агломеративные алгоритмы начинают с каждого элемента как отдельного кластера и объединяют их в последовательно более крупные кластеры. Алгоритмы разделения начинают со всего набора и продолжают делить его на последовательно меньшие кластеры. Иерархическая кластеризация рассчитывается с использованием метрик евклидовых пространств , наиболее часто используемым является евклидово расстояние, вычисляемое путем нахождения квадрата разницы между каждой переменной, сложения всех квадратов и нахождения квадратного корня из указанной суммы. Примером алгоритма иерархической кластеризации является BIRCH , который особенно хорош в биоинформатике из-за его почти линейной временной сложности, учитывая, как правило, большие наборы данных. [27] Алгоритмы секционирования основаны на указании начального количества групп и итеративном перераспределении объектов между группами для обеспечения сходимости. Этот алгоритм обычно определяет все кластеры одновременно. Большинство приложений используют один из двух популярных эвристических методов: алгоритм k-средних или k-medoids . , не требуют начального количества групп Другие алгоритмы, такие как распространение аффинности . В геномике этот алгоритм использовался как для кластеризации кластеров биосинтетических генов в семействах генных кластеров (GCF), так и для кластеризации указанных GCF. [28]
Рабочий процесс [ править ]
Обычно рабочий процесс применения машинного обучения к биологическим данным состоит из четырех этапов: [2]
- Запись, включая захват и хранение. На этом этапе различные источники информации могут быть объединены в один набор.
- Предварительная обработка, включая очистку и реструктуризацию в готовую для анализа форму. На этом этапе неисправленные данные исключаются или исправляются, а отсутствующие данные могут быть условно рассчитаны и выбраны соответствующие переменные.
- Анализ, оценка данных с использованием контролируемых или неконтролируемых алгоритмов. Алгоритм обычно обучается на подмножестве данных, оптимизирует параметры и оценивается на отдельном подмножестве тестов.
- Визуализация и интерпретация, при которой знания эффективно представляются с использованием различных методов для оценки значимости и важности результатов.
Ошибки данных [ править ]
- Дублирование данных является серьезной проблемой в биоинформатике. Общедоступные данные могут иметь неопределенное качество. [29]
- Ошибки во время экспериментов. [29]
- Ошибочная интерпретация. [29]
- Ошибки при наборе текста. [29]
- В экспериментах используются нестандартизированные методы (3D-структура в PDB из нескольких источников, рентгеновская дифракция, теоретическое моделирование, ядерный магнитный резонанс и др.). [29]
Приложения [ править ]
В общем, систему машинного обучения обычно можно обучить распознавать элементы определенного класса при наличии достаточных выборок. [30] Например, методы машинного обучения можно обучить распознавать определенные визуальные особенности, такие как места сращивания. [31]
Машины опорных векторов широко используются в геномных исследованиях рака. [32] Кроме того, глубокое обучение включено в биоинформационные алгоритмы. Приложения глубокого обучения использовались для регуляторной геномики и визуализации клеток. [33] Другие приложения включают классификацию медицинских изображений, анализ геномных последовательностей, а также классификацию и прогнозирование структуры белков. [34] Глубокое обучение применялось к регуляторной геномике, определению вариантов и оценке патогенности. [35] Обработка естественного языка и анализ текста помогли понять такие явления, как белок-белковое взаимодействие, взаимосвязь генов и заболеваний, а также предсказать структуры и функции биомолекул. [36]
Прецизионная/персонализированная медицина [ править ]
обработки естественного языка Алгоритмы персонализировали медицину для пациентов, страдающих генетическими заболеваниями, путем объединения извлечения клинической информации и геномных данных, доступных от пациентов. Такие институты, как Сеть исследований фармакогеномики, финансируемая здравоохранением, занимаются поиском методов лечения рака молочной железы. [37]
Точная медицина учитывает индивидуальную геномную изменчивость, обеспечиваемую крупномасштабными биологическими базами данных. Машинное обучение может применяться для выполнения функции сопоставления между (группами пациентов) и конкретными методами лечения. [38]
Вычислительные методы используются для решения других проблем, таких как эффективный дизайн праймеров для ПЦР , анализ биологических изображений и обратная трансляция белков (что, учитывая вырождение генетического кода, представляет собой сложную комбинаторную задачу). [2]
Геномика [ править ]
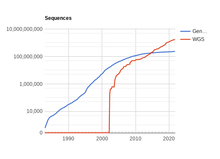
Хотя данные о геномных последовательностях исторически были скудными из-за технической сложности секвенирования фрагмента ДНК, количество доступных последовательностей растет. В среднем количество баз , доступных в публичном репозитории GenBank, удваивалось каждые 18 месяцев с 1982 года. [39] Однако, хотя необработанные данные становились все более доступными и доступными, по состоянию на 2002 г. [update]Биологическая интерпретация этих данных происходила гораздо медленнее. [40] Это привело к растущей потребности в разработке инструментов вычислительной геномики , включая системы машинного обучения, которые могут автоматически определять расположение генов, кодирующих белки, внутри заданной последовательности ДНК (т.е. предсказание генов ). [40]
Предсказание генов обычно выполняется посредством как внешнего, так и внутреннего поиска . [40] Для внешнего поиска входная последовательность ДНК прогоняется через большую базу данных последовательностей, гены которых были ранее обнаружены, и их местоположения аннотированы, а также идентифицируются гены целевой последовательности путем определения того, какие строки оснований в последовательности гомологичны известным последовательностям генов. Однако не все гены в данной входной последовательности можно идентифицировать только по гомологии из-за ограничений размера базы данных известных и аннотированных последовательностей генов. Следовательно, необходим внутренний поиск, когда программа прогнозирования генов пытается идентифицировать оставшиеся гены только по последовательности ДНК. [40]
Машинное обучение также использовалось для решения проблемы множественного выравнивания последовательностей , которая включает в себя выравнивание многих последовательностей ДНК или аминокислот для определения областей сходства, которые могут указывать на общую историю эволюции. [2] Его также можно использовать для обнаружения и визуализации перестроек генома. [41]
Протеомика [ править ]

Белки , цепочки аминокислот , получают большую часть своих функций за счет сворачивания белка , при котором они образуют трехмерную структуру, включая первичную структуру , вторичную структуру ( альфа-спирали и бета-листы ), третичную структуру и четвертичную структуру. .
Прогнозирование вторичной структуры белка является основным направлением этой области, поскольку третичная и четвертичная структуры определяются на основе вторичной структуры. [4] Определение истинной структуры белка является дорогостоящим и трудоемким занятием, что увеличивает потребность в системах, которые могут точно предсказать структуру белка путем непосредственного анализа аминокислотной последовательности. [4] [2] До появления машинного обучения исследователям приходилось делать прогнозы вручную. Эта тенденция началась в 1951 году, когда Полинг и Кори опубликовали свою работу по предсказанию конфигурации водородных связей белка по полипептидной цепи. [42] Автоматическое обучение функциям достигает точности 82-84%. [4] [43] Современный уровень прогнозирования вторичной структуры использует систему под названием DeepCNF (глубокие сверточные нейронные поля), которая опирается на модель машинного обучения искусственных нейронных сетей для достижения точности примерно 84% при задаче классификации аминокислот. белковой последовательности в один из трех структурных классов (спираль, лист или клубок). [43] [ нужно обновить ] Теоретический предел трехуровневой вторичной структуры белка составляет 88–90%. [4]
Машинное обучение также применялось для решения проблем протеомики, таких как предсказание боковой цепи белка , моделирование белковой петли и предсказание карты контактов белков . [2]
Метагеномика [ править ]
Метагеномика — это изучение микробных сообществ из образцов ДНК из окружающей среды. [44] В настоящее время при внедрении инструментов машинного обучения преобладают ограничения и проблемы из-за большого количества данных в образцах окружающей среды. [45] Суперкомпьютеры и веб-серверы облегчили доступ к этим инструментам. [46] Высокая размерность наборов данных о микробиоме является серьезной проблемой при изучении микробиома; это существенно ограничивает возможности нынешних подходов к выявлению истинных различий и увеличивает вероятность ложных открытий. [47] [ нужен лучший источник ]
Несмотря на свою важность, инструменты машинного обучения, связанные с метагеномикой, сосредоточены на изучении микробиоты кишечника и взаимосвязи с заболеваниями пищеварительной системы, такими как воспалительные заболевания кишечника (ВЗК), инфекция Clostridioides difficile (CDI), колоректальный рак и диабет , в поисках лучшей диагностики и методы лечения. [46] Было разработано множество алгоритмов для классификации микробных сообществ в соответствии с состоянием здоровья хозяина, независимо от типа данных о последовательности, например, 16S рРНК или полногеномное секвенирование (WGS), с использованием таких методов, как наименьшее абсолютное сокращение и классификатор операторов отбора, случайный лес , модель контролируемой классификации и модель дерева с градиентным усилением. нейронные сети , такие как рекуррентные нейронные сети (RNN), сверточные нейронные сети (CNN) и нейронные сети Хопфилда . Были добавлены [46] Например, в 2018 году Фиораванти и др. разработал алгоритм под названием Ph-CNN для классификации образцов данных от здоровых пациентов и пациентов с симптомами ВЗК (чтобы различать здоровых и больных пациентов) с использованием филогенетических деревьев и сверточных нейронных сетей. [48]
Кроме того, методы случайного леса (RF) и реализованные меры важности помогают идентифицировать виды микробиома, которые можно использовать для различения больных и здоровых образцов. Однако производительность дерева решений и разнообразие деревьев решений в ансамбле существенно влияют на производительность RF-алгоритмов. Ошибка обобщения для RF измеряет точность отдельных классификаторов и их взаимозависимость. Таким образом, проблемы высокой размерности наборов данных о микробиомах создают проблемы. Эффективные подходы требуют множества возможных комбинаций переменных, что экспоненциально увеличивает вычислительную нагрузку по мере увеличения количества функций. [47]
Для анализа микробиома в 2020 году Данг и Кишино [47] разработал новый конвейер анализа. Ядром конвейера является радиочастотный классификатор в сочетании с механизмом выбора переменных пересылки (RF-FVS), который выбирает основной набор микробных видов или функциональных сигнатур минимального размера, которые максимизируют производительность прогнозирующего классификатора. Фреймворк сочетает в себе:
- идентификация нескольких важных особенностей с помощью массового параллельного прямого выбора переменных процедуры
- картирование выбранных видов на филогенетическом дереве и
- прогнозирование функциональных профилей с помощью анализа функционального обогащения генов на основе метагеномных данных 16S рРНК .
Они продемонстрировали эффективность, проанализировав два опубликованных набора данных из крупномасштабных исследований «случай-контроль»:
- Данные ампликона гена 16S рРНК при инфекции C. difficile (CDI) и
- Данные дробовой метагеномики колоректального рака человека (КРР).
Предложенный подход повысил точность с 81% до 99,01% для CDI и с 75,14% до 90,17% для CRC.
Использование машинного обучения в образцах окружающей среды изучено меньше, возможно, из-за сложности данных, особенно из WGS. Некоторые работы показывают, что эти инструменты можно применять к образцам окружающей среды. В 2021 году Дхунгель и др., [49] разработал пакет R под названием MegaR. Этот пакет позволяет работать с 16S рРНК и целыми метагеномными последовательностями для создания таксономических профилей и моделей классификации с помощью моделей машинного обучения. MegaR включает удобную среду визуализации для улучшения пользовательского опыта. Машинное обучение в метагеномике окружающей среды может помочь ответить на вопросы, связанные с взаимодействием между микробными сообществами и экосистемами, например, работа Сюня и др. в 2021 году. [50] где использование различных методов машинного обучения позволило получить представление о взаимосвязи между почвой, биоразнообразием микробиома и стабильностью экосистемы.
Микрочипы [ править ]
Микрочипы , разновидность лаборатории на чипе , используются для автоматического сбора данных о больших объемах биологического материала. Машинное обучение может помочь в анализе и применяется для идентификации, классификации и индукции генетических сетей. [2]
Эта технология особенно полезна для мониторинга экспрессии генов, помогая диагностировать рак путем изучения того, какие гены экспрессируются. [51] Одна из основных задач — на основе собранных данных определить, какие гены экспрессируются. [2] Кроме того, из-за огромного количества генов, данные о которых собираются с помощью микрочипа, отсеивание большого количества нерелевантных данных для задачи идентификации экспрессируемых генов является сложной задачей. Машинное обучение представляет собой потенциальное решение, поскольку для выполнения этой идентификации можно использовать различные методы классификации. Наиболее часто используемые методы — это сети с радиальными базисными функциями , глубокое обучение , байесовская классификация , деревья решений и случайный лес . [51]
Системная биология
Системная биология фокусируется на изучении поведения, возникающего в результате сложных взаимодействий простых биологических компонентов в системе. Такие компоненты могут включать ДНК, РНК, белки и метаболиты. [52]
Машинное обучение использовалось для моделирования этих взаимодействий в таких областях, как генетические сети, сети передачи сигналов и метаболические пути. [2] Вероятностные графические модели — метод машинного обучения для определения взаимосвязей между различными переменными — являются одним из наиболее часто используемых методов моделирования генетических сетей. [2] Кроме того, машинное обучение применялось для решения задач системной биологии, таких как идентификация сайтов связывания факторов транскрипции с использованием оптимизации цепи Маркова . [2] Генетические алгоритмы , методы машинного обучения, основанные на естественном процессе эволюции, использовались для моделирования генетических сетей и регуляторных структур. [2]
Другие приложения машинного обучения в системной биологии включают задачу прогнозирования функций ферментов, высокопроизводительный анализ данных микрочипов, анализ полногеномных ассоциативных исследований для лучшего понимания маркеров заболеваний, прогнозирование функций белков. [53]
Эволюция [ править ]
Эта область, в частности реконструкция филогенетического дерева , использует особенности методов машинного обучения. Филогенетические деревья представляют собой схематическое изображение эволюции организмов. Первоначально они были построены с использованием таких особенностей, как морфологические и метаболические особенности. Позже, благодаря доступности геномных последовательностей, при построении алгоритма филогенетического дерева была использована концепция, основанная на сравнении геномов. С помощью методов оптимизации сравнение было проведено посредством множественного выравнивания последовательностей. [54]
Диагностика инсульта [ править ]
Методы машинного обучения для анализа данных нейровизуализации используются для диагностики инсульта . Исторически сложилось так, что несколько подходов к этой проблеме включали использование нейронных сетей. [55] [56]
Несколько подходов к обнаружению инсультов используют машинное обучение. По предложению Мирцхулавы, [57] Сети прямой связи были протестированы для обнаружения инсультов с использованием нейронной визуализации. По предложению Титано [58] Методы 3D-CNN были протестированы в контролируемой классификации для проверки КТ-изображений головы на предмет острых неврологических событий. трехмерные методы CNN и SVM . Часто используются [56]
Анализ текста [ править ]
Увеличение биологических публикаций увеличило сложность поиска и обобщения актуальной доступной информации по заданной теме. Эта задача известна как извлечение знаний . Это необходимо для сбора биологических данных, которые затем можно будет использовать в алгоритмах машинного обучения для создания новых биологических знаний. [2] [59] Для этой задачи извлечения знаний можно использовать машинное обучение с использованием таких методов, как обработка естественного языка, для извлечения полезной информации из отчетов, созданных человеком, в базе данных. Text Nailing , альтернативный подход к машинному обучению, способный извлекать особенности из клинических заметок, был представлен в 2017 году.
Этот метод применялся для поиска новых мишеней для лекарств, поскольку эта задача требует изучения информации, хранящейся в биологических базах данных и журналах. [59] Аннотации белков в базах данных белков часто не отражают полный известный набор знаний о каждом белке, поэтому дополнительную информацию необходимо извлекать из биомедицинской литературы. Машинное обучение применялось для автоматического аннотирования функций генов и белков, определения субклеточной локализации белков , анализа массива экспрессии ДНК , крупномасштабного анализа взаимодействия белков и анализа взаимодействия молекул. [59]
Еще одним применением интеллектуального анализа текста является обнаружение и визуализация отдельных участков ДНК при наличии достаточных справочных данных. [60]
численности кластеров биосинтетических Кластеризация и профилирование генов
Микробные сообщества – это сложные совокупности разнообразных микроорганизмов. [61] где партнеры-симбионты постоянно продуцируют разнообразные метаболиты, полученные в результате первичного и вторичного (специализированного) метаболизма, из которых метаболизм играет важную роль в микробном взаимодействии. [62] Метагеномные и метатранскриптомные данные являются важным источником для расшифровки коммуникационных сигналов.
Молекулярные механизмы производят специализированные метаболиты различными способами. Биосинтетические генные кластеры (БГК) привлекают внимание, поскольку некоторые метаболиты являются клинически ценными, антимикробными, противогрибковыми, противопаразитарными, противоопухолевыми и иммуносупрессивными агентами, вырабатываемыми в результате модульного действия мультиферментативных, мультидоменных кластеров генов. такие как нерибосомальные пептидсинтазы (NRPS) и поликетидсинтазы (PKS). [63] Разнообразные исследования [64] [65] [66] [67] [68] [69] [70] [71] показывают, что группировка BGC, которые имеют гомологичные основные гены, в семейства генных кластеров (GCF), может дать полезную информацию о химическом разнообразии анализируемых штаммов и может способствовать связыванию BGC с их вторичными метаболитами. [65] [67] GCF использовались в качестве функциональных маркеров в исследованиях здоровья человека. [72] [73] и изучить способность почвы подавлять грибковые патогены. [74] Учитывая их прямую связь с каталитическими ферментами и соединениями, образующимися в результате кодируемых ими путей, BGC/GCF могут служить показателем для изучения химического пространства микробного вторичного метаболизма. Каталогизация GCF в секвенированных микробных геномах дает представление о существующем химическом разнообразии и дает представление о будущих приоритетах. [64] [66] Такие инструменты, как BiG-SLiCE и BIG-MAP. [75] появились с единственной целью раскрыть важность BGC в естественной среде.
РиПП Расшифровка строения химического
Увеличение числа экспериментально охарактеризованных рибосомально синтезируемых и посттрансляционно модифицированных пептидов (RiPP), а также доступность информации об их последовательности и химической структуре, выбранной из таких баз данных, как BAGEL, BACTIBASE, MIBIG и THIOBASE, дают возможность разработать машины средства обучения для расшифровки химической структуры и их классификации.
В 2017 году исследователи из Национального института иммунологии Нью-Дели, Индия, разработали RiPPMiner. [76] программное обеспечение, биоинформатический ресурс для декодирования химических структур RiPP путем анализа генома. Веб-сервер RiPPMiner состоит из интерфейса запросов и базы данных RiPPDB. RiPPMiner определяет 12 подклассов RiPP, предсказывая место расщепления лидерного пептида и окончательную сшивку химической структуры RiPP.
сходства Оценка масс - спектрального
Многие метаболомные исследования, основанные на тандемной масс-спектрометрии ( МС/МС ) , такие как сопоставление библиотек и создание молекулярных сетей, используют спектральное сходство в качестве показателя структурного сходства. Спец2век [77] Алгоритм предоставляет новый способ оценки спектрального сходства, основанный на Word2Vec . Spec2Vec изучает фрагментарные отношения в большом наборе спектральных данных, чтобы оценить спектральное сходство между молекулами и классифицировать неизвестные молекулы посредством этих сравнений.
Для системной аннотации некоторые метаболомические исследования полагаются на сопоставление измеренных масс-спектров фрагментации с библиотечными спектрами или контрастирование спектров с помощью сетевого анализа. Оценочные функции используются для определения сходства между парами спектров фрагментов в рамках этих процессов. До сих пор ни одно исследование не предложило оценки, которые значительно отличались бы от обычно используемого сходства на основе косинуса . [78]
Базы данных [ править ]
Важной частью биоинформатики является управление большими наборами данных, известными как справочные базы данных. Базы данных существуют для каждого типа биологических данных, например, для кластеров биосинтетических генов и метагеномов.
Общие базы данных по биоинформатике [ править ]
биотехнологической информации Национальный центр
Национальный центр биотехнологической информации (NCBI) [79] предоставляет большой набор онлайн-ресурсов для биологической информации и данных, включая GenBank базу данных последовательностей нуклеиновых кислот PubMed и базу данных цитат и рефератов для опубликованных журналов по биологическим наукам. Многие веб-приложения дополняются специальными реализациями программы BLAST, оптимизированными для поиска специализированных наборов данных. Ресурсы включают управление данными PubMed, функциональные элементы RefSeq, загрузку данных генома, API служб вариаций, Magic-BLAST, QuickBLASTp и идентичные группы белков. Доступ ко всем этим ресурсам можно получить через NCBI. [80]
анализ кластеров биосинтетических Биоинформатический генов
антиСМЭШ [ править ]
antiSMASH позволяет быстро идентифицировать, аннотировать и анализировать кластеры генов биосинтеза вторичных метаболитов в геномах бактерий и грибов. Он интегрируется и взаимодействует с большим количеством инструментов анализа вторичных метаболитов in silico . [81]
гутСМЭШ [ править ]
GutSMASH — это инструмент, который систематически оценивает метаболический потенциал бактерий путем прогнозирования как известных, так и новых кластеров анаэробных кишечника метаболических генов (MGC) на основе микробиома .
МИБиГ [ править ]
МИБиГ, [82] минимальная информация о спецификации кластера биосинтетических генов обеспечивает стандарт для аннотаций и метаданных о кластерах биосинтетических генов и их молекулярных продуктах. MIBIG — это проект Консорциума по геномным стандартам, который основан на структуре минимальной информации о любой последовательности (MIxS). [83]
MIBiG облегчает стандартизированное хранение и извлечение данных о кластерах биосинтетических генов, а также разработку комплексных инструментов сравнительного анализа. Это расширяет возможности исследований следующего поколения в области биосинтеза, химии и экологии широких классов социально значимых биоактивных вторичных метаболитов , основанных на надежных экспериментальных данных и богатых компонентах метаданных. [84]
SILVA[editСИЛЬВА
СИЛЬВА [85] это междисциплинарный проект биологов и компьютерщиков, собирающий полную базу данных рибосомальных (рРНК) последовательностей генов, как малых ( 16S , 18S , SSU), так и больших ( 23S , 28S , LSU) субъединиц, принадлежащих бактериям, археям. и домены эукариев. Эти данные находятся в свободном доступе для академического и коммерческого использования. [86]
Зеленые гены [ править ]
Зеленые гены [87] представляет собой полноразмерную базу данных генов 16S рРНК , которая обеспечивает скрининг химер, стандартное выравнивание и тщательно подобранную таксономию, основанную на выводе дерева de novo. [88] [89] Обзор:
- 1 012 863 последовательности РНК из 92 684 организмов внесли вклад в RNAcentral.
- Самая короткая последовательность состоит из 1253 нуклеотидов, самая длинная — 2368.
- Средняя длина составляет 1402 нуклеотида.
- Версия базы данных: 13.5.
Древа таксономия Открытая Жизни
Таксономия Открытого Древа Жизни (OTT) [90] Целью проекта является создание полного, динамичного и доступного в цифровом формате Древа жизни путем синтеза опубликованных филогенетических деревьев вместе с таксономическими данными. Филогенетические деревья были классифицированы, выровнены и объединены. Таксономии использовались для заполнения редких регионов и пробелов, оставленных филогениями. OTT представляет собой базу, которая мало использовалась для анализа секвенирования региона 16S, однако она имеет большее количество последовательностей, классифицированных таксономически вплоть до уровня рода, по сравнению с SILVA и Greengenes. Однако с точки зрения классификации на краевом уровне он содержит меньший объем информации. [91]
рибосомальной Проект данных базы
Проект рибосомальной базы данных (RDP) [92] представляет собой базу данных, которая содержит рибосомальные последовательности РНК (рРНК) малых субъединиц бактериального и архейного домена ( 16S ); и последовательности грибковых рРНК больших субъединиц ( 28S ). [93]
Ссылки [ править ]
- ^ Чикко Д. (декабрь 2017 г.). «Десять быстрых советов по машинному обучению в вычислительной биологии» . Добыча биоданных . 10 (35): 35. дои : 10.1186/s13040-017-0155-3 . ПМК 5721660 . ПМИД 29234465 .
- ↑ Перейти обратно: Перейти обратно: а б с д и ж г час я дж к л м Ларраньяга П., Кальво Б., Сантана Р., Бьелза С., Галдиано Дж., Инса И. и др. (март 2006 г.). «Машинное обучение в биоинформатике» . Брифинги по биоинформатике . 7 (1): 86–112. дои : 10.1093/нагрудник/bbk007 . ПМИД 16761367 .
- ^ Перес-Вольфейл Э., Торреноа О., Беллис Л.Дж., Фернандес П.Л., Лескосек Б., Треллеса О. (декабрь 2018 г.). «Подготовка биоинформатиков в области высокопроизводительных вычислений» . Гелион . 4 (12): e01057. Бибкод : 2018Heliy...401057P . дои : 10.1016/j.heliyon.2018.e01057 . ПМК 6299036 . ПМИД 30582061 .
- ↑ Перейти обратно: Перейти обратно: а б с д и Ян Ю, Гао Дж, Ван Дж, Хеффернан Р, Хансон Дж, Паливал К, Чжоу Ю (май 2018 г.). «Шестьдесят пять лет долгого пути в предсказании вторичной структуры белка: последний этап?» . Брифинги по биоинформатике . 19 (3): 482–494. дои : 10.1093/нагрудник/bbw129 . ПМЦ 5952956 . ПМИД 28040746 .
- ^ Шастри К.А., Санджай Х.А. (2020). «Машинное обучение для биоинформатики» . Шриниваса К., Сиддеш Г., Манисехар С. (ред.). Принципы статистического моделирования и машинного обучения для методов, инструментов и приложений биоинформатики . Алгоритмы интеллектуальных систем. Сингапур: Спрингер. стр. 25–39. дои : 10.1007/978-981-15-2445-5_3 . ISBN 978-981-15-2445-5 . S2CID 214350490 . Проверено 28 июня 2021 г.
- ↑ Перейти обратно: Перейти обратно: а б с Суэйдан Х., Никольски М. (2019). «Машинное обучение для метагеномики: методы и инструменты». Метагеномика . 1 . arXiv : 1510.06621 . дои : 10.1515/metgen-2016-0001 . ISSN 2449-7657 . S2CID 17418188 .
- ^ Рабинер Л., Хуанг Б. (январь 1986 г.). «Введение в скрытые модели Маркова» . Журнал IEEE ASSP . 3 (1): 4–16. дои : 10.1109/MASSP.1986.1165342 . ISSN 1558-1284 . S2CID 11358505 .
- ^ Джексон Ч., Шарплс Л.Д., Томпсон С.Г., Даффи С.В., Коуто Э. (июль 2003 г.). «Многогосударственные марковские модели прогрессирования заболевания с ошибкой классификации». Журнал Королевского статистического общества, серия D (Статист) . 52 (2): 193–209. дои : 10.1111/1467-9884.00351 . S2CID 9824404 .
- ^ Аморос Р., Кинг Р., Тойода Х., Кумада Т., Джонсон П.Дж., Берд Т.Г. (30 мая 2019 г.). «Скрытая марковская модель непрерывного времени для наблюдения за раком с использованием сывороточных биомаркеров с применением к гепатоцеллюлярной карциноме» . Метрон . 77 (2): 67–86. дои : 10.1007/s40300-019-00151-8 . ПМК 6820468 . ПМИД 31708595 .
- ^ Эдди С.Р. (1 октября 1998 г.). «Профиль скрытых марковских моделей» . Биоинформатика . 14 (9): 755–63. дои : 10.1093/биоинформатика/14.9.755 . ПМИД 9918945 .
- ^ МакКлинток Б.Т., Лангрок Р., Хименес О., Кэм Э., Борчерс Д.Л., Гленни Р., Паттерсон Т.А. (декабрь 2020 г.). «Раскрытие динамики экологического состояния с помощью скрытых марковских моделей» . Экологические письма . 23 (12): 1878–1903. arXiv : 2002.10497 . Бибкод : 2020EcolL..23.1878M . дои : 10.1111/ele.13610 . ПМК 7702077 . ПМИД 33073921 .
- ^ Чжан В (1988). «Сдвиг-инвариантная нейронная сеть распознавания образов и ее оптическая архитектура» . Материалы ежегодной конференции Японского общества прикладной физики .
- ^ Чжан В., Ито К., Танида Дж., Ичиока Ю. (ноябрь 1990 г.). «Модель параллельной распределенной обработки с локальными пространственно-инвариантными соединениями и ее оптическая архитектура». Прикладная оптика . 29 (32): 4790–7. Бибкод : 1990ApOpt..29.4790Z . дои : 10.1364/AO.29.004790 . ПМИД 20577468 .
- ↑ Перейти обратно: Перейти обратно: а б Бишоп Кристофер М. (17 августа 2006 г.). Распознавание образов и машинное обучение . Нью-Йорк: Спрингер. ISBN 978-0-387-31073-2 .
- ^ Фукусима К (2007 г.). «Неокогнитрон» . Схоларпедия . 2 (1): 1717. Бибкод : 2007SchpJ...2.1717F . doi : 10.4249/scholarpedia.1717 .
- ^ Хьюбель Д.Х., Визель Т.Н. (март 1968 г.). «Рецептивные поля и функциональная архитектура полосатой коры обезьян» . Журнал физиологии . 195 (1): 215–43. doi : 10.1113/jphysicalol.1968.sp008455 . ПМЦ 1557912 . ПМИД 4966457 .
- ^ Фукусима К (1980). «Неокогнитрон: самоорганизующаяся модель нейронной сети для механизма распознавания образов, на который не влияет сдвиг положения». Биологическая кибернетика . 36 (4): 193–202. дои : 10.1007/BF00344251 . ПМИД 7370364 . S2CID 206775608 .
- ^ Мацугу М., Мори К., Митари Ю., Канеда Ю. (2003). «Субъектное независимое распознавание выражения лица с надежным обнаружением лиц с использованием сверточной нейронной сети». Нейронные сети . 16 (5–6): 555–9. дои : 10.1016/S0893-6080(03)00115-1 . ПМИД 12850007 .
- ^ Фиораванти Д., Джарратано И., Маджио В., Агостинелли С., Кьеричи М., Юрман Г., Фурланелло С. (март 2018 г.). «Филогенетические сверточные нейронные сети в метагеномике» . БМК Биоинформатика . 19 (Приложение 2): 49. doi : 10.1186/s12859-018-2033-5 . ПМЦ 5850953 . ПМИД 29536822 .
- ^ Цзи, Янжун; Чжоу, Чжихан; Лю, Хан; Давулури, Рамана V (9 августа 2021 г.). Келсо, Джанет (ред.). «DNABERT: предварительно обученные представления двунаправленного кодировщика из модели Transformers для языка ДНК в геноме» . Биоинформатика . 37 (15): 2112–2120. doi : 10.1093/биоинформатика/btab083 . ISSN 1367-4803 . ПМЦ 11025658 . ПМИД 33538820 .
- ^ Гюндюз, Хусейн Анил; Биндер, Мартин; То, Сяо-Инь; Мрешес, Рене; Бишль, Бернд; Макхарди, Элис К.; Мюнх, Филипп К.; Резаи, Мина (11 сентября 2023 г.). «Метод глубокого обучения с самоконтролем для эффективного обучения геномике с использованием данных» . Коммуникационная биология . 6 (1): 928. дои : 10.1038/s42003-023-05310-2 . ISSN 2399-3642 . ПМЦ 10495322 . ПМИД 37696966 .
- ^ Хо ТК (1995). Леса случайных решений . Материалы 3-й Международной конференции по анализу и распознаванию документов, Монреаль, Квебек, 14–16 августа 1995 г., стр. 278–282.
- ^ Дитерих Т. (2000). Экспериментальное сравнение трех методов построения ансамблей деревьев решений: пакетирование, повышение и рандомизация . Академическое издательство Клувер. стр. 139–157.
- ^ Брейман, Лео (2001). «Радомские леса» . Машинное обучение . 45 (1): 5–32. Бибкод : 2001MachL..45....5B . дои : 10.1023/А:1010933404324 . S2CID 89141 .
- ↑ Перейти обратно: Перейти обратно: а б Чжан С., Ма Ю (2012). Ансамбльное машинное обучение: методы и приложения . Нью-Йорк: Springer New York Dordrecht Heidelberg London. стр. 157–175. ISBN 978-1-4419-9325-0 .
- ^ Карим М.Р., Беян О., Заппа А., Коста И.Г., Ребхольц-Шуман Д., Кочес М., Декер С. (январь 2021 г.). «Подходы к кластеризации на основе глубокого обучения для биоинформатики» . Брифинги по биоинформатике . 22 (1): 393–415. дои : 10.1093/нагрудник/bbz170 . ПМЦ 7820885 . ПМИД 32008043 .
- ^ Лорбер Б., Косарева А., Дева Б., Софтич Д., Руппель П., Куппер А. (1 марта 2018 г.). «Вариации алгоритма кластеризации BIRCH» . Исследование больших данных . 11 : 44–53. дои : 10.1016/j.bdr.2017.09.002 .
- ^ Наварро-Муньос Х.К., Селем-Мохика Н., Маллоуни М.В., Каутсар С.А., Трайон Дж.Х., Паркинсон Э.И. и др. (январь 2020 г.). «Вычислительная система для изучения крупномасштабного биосинтетического разнообразия» . Химическая биология природы . 16 (1): 60–68. дои : 10.1038/s41589-019-0400-9 . ПМЦ 6917865 . ПМИД 31768033 .
- ↑ Перейти обратно: Перейти обратно: а б с д и Шастри К.А., Санджай Х.А. (2020). «Машинное обучение для биоинформатики». Принципы статистического моделирования и машинного обучения для методов, инструментов и приложений биоинформатики . Алгоритмы интеллектуальных систем. Спрингер Сингапур. стр. 25–39. дои : 10.1007/978-981-15-2445-5_3 . ISBN 978-981-15-2444-8 . S2CID 214350490 .
- ^ Либбрехт М.В., Нобл В.С. (июнь 2015 г.). «Приложения машинного обучения в генетике и геномике» . Обзоры природы. Генетика . 16 (6): 321–32. дои : 10.1038/nrg3920 . ПМК 5204302 . ПМИД 25948244 .
- ^ Дегроев С., Де Баетс Б., Ван де Пер Ю., Рузе П. (2002). «Выбор подмножества функций для прогнозирования места сращивания» . Биоинформатика . 18 (Приложение 2): С75-83. doi : 10.1093/bioinformatics/18.suppl_2.s75 . ПМИД 12385987 .
- ^ Хуан С., Цай Н., Пачеко П.П., Наррандес С., Ван Ю, Сюй В. (январь 2018 г.). «Применение машинного обучения опорных векторов (SVM) в геномике рака» . Геномика и протеомика рака . 15 (1): 41–51. дои : 10.21873/cgp.20063 . ПМЦ 5822181 . ПМИД 29275361 .
- ^ Ангермюллер К., Пярнамаа Т., Партс L, Стегле О (июль 2016 г.). «Глубокое обучение для вычислительной биологии» . Молекулярная системная биология . 12 (7): 878. doi : 10.15252/msb.20156651 . ПМЦ 4965871 . ПМИД 27474269 .
- ^ Цао С., Лю Ф., Тан Х., Сун Д., Шу В., Ли В. и др. (февраль 2018 г.). «Глубокое обучение и его применение в биомедицине» . Геномика, протеомика и биоинформатика . 16 (1): 17–32. дои : 10.1016/j.gpb.2017.07.003 . ПМК 6000200 . ПМИД 29522900 .
- ^ Зоу Дж., Хусс М., Абид А., Мохаммади П., Торкамани А., Теленти А. (январь 2019 г.). «Букварь по глубокому изучению геномики». Природная генетика . 51 (1): 12–18. дои : 10.1038/s41588-018-0295-5 . ПМИД 30478442 . S2CID 205572042 .
- ^ Цзэн Цзи, Ши Х, Ву Ю, Хун Цзи (2015). «Обзор методов обработки естественного языка в биоинформатике» . Вычислительные и математические методы в медицине . 2015 (D1): 674296. doi : 10.1155/2015/674296 . ПМЦ 4615216 . ПМИД 26525745 .
- ^ Цзэн З, Ши Х, Ву Ю, Хун З (2012). «Обзор методов обработки естественного языка в биоинформатике» . Вычислительные и математические методы в медицине . 2015 (D1): 674296. doi : 10.1016/B978-0-12-385467-4.00006-3 . ПМЦ 4615216 . ПМИД 26525745 .
- ^ Цзэн Цзи, Ши Х, Ву Ю, Хун Цзи (2017). «Обзор методов обработки естественного языка в биоинформатике» . Вычислительные и математические методы в медицине . 2015 (D1): 674296. doi : 10.1155/2015/674296 . ПМЦ 4615216 . ПМИД 26525745 .
- ^ «Статистика Генбанка и WGS» . www.ncbi.nlm.nih.gov . Проверено 25 ноября 2023 г.
- ↑ Перейти обратно: Перейти обратно: а б с д Мате С., Саго М.Ф., Шиекс Т., Рузе П. (октябрь 2002 г.). «Современные методы генного прогнозирования, их сильные и слабые стороны» . Исследования нуклеиновых кислот . 30 (19): 4103–17. дои : 10.1093/nar/gkf543 . ПМК 140543 . ПМИД 12364589 .
- ^ Пратас Д., Силва Р.М., Пиньо А.Дж., Феррейра П.Дж. (май 2015 г.). «Метод без выравнивания для поиска и визуализации перестроек между парами последовательностей ДНК» . Научные отчеты . 5 (10203): 10203. Бибкод : 2015NatSR...510203P . дои : 10.1038/srep10203 . ПМЦ 4434998 . ПМИД 25984837 .
- ^ Полинг Л., Кори Р.Б., Брэнсон Х.Р. (апрель 1951 г.). «Строение белков; две спиральные конфигурации полипептидной цепи с водородными связями» . Труды Национальной академии наук Соединенных Штатов Америки . 37 (4): 205–11. Бибкод : 1951ПНАС...37..205П . дои : 10.1073/pnas.37.4.205 . ПМЦ 1063337 . ПМИД 14816373 .
- ↑ Перейти обратно: Перейти обратно: а б Ван С., Пэн Дж., Ма Дж., Сюй Дж. (январь 2016 г.). «Прогнозирование вторичной структуры белка с использованием глубоких сверточных нейронных полей» . Научные отчеты . 6 : 18962. arXiv : 1512.00843 . Бибкод : 2016NatSR...618962W . дои : 10.1038/srep18962 . ПМЦ 4707437 . ПМИД 26752681 .
- ^ Ризенфельд К.С., Шлосс П.Д., Хандельсман Дж. (2004). «Метагеномика: геномный анализ микробных сообществ». Ежегодный обзор генетики . 38 (1): 525–52. дои : 10.1146/annurev.genet.38.072902.091216 . ПМИД 15568985 .
- ^ Суэйдан, Хайссам; Никольский, Маха (1 января 2017 г.). «Машинное обучение для метагеномики: методы и инструменты» . Метагеномика . 1 (1). arXiv : 1510.06621 . дои : 10.1515/metgen-2016-0001 . ISSN 2449-7657 . S2CID 17418188 .
- ↑ Перейти обратно: Перейти обратно: а б с Линь Ю, Ван Джи, Ю Дж, Сун Джей Джей (апрель 2021 г.). «Искусственный интеллект и метагеномика при кишечных заболеваниях» . Журнал гастроэнтерологии и гепатологии . 36 (4): 841–847. дои : 10.1111/jgh.15501 . ПМИД 33880764 . S2CID 233312307 .
- ↑ Перейти обратно: Перейти обратно: а б с Данг Т., Кишино Х. (январь 2020 г.). «Обнаружение важных компонентов микробиомов с помощью случайного леса с прямым отбором переменных и филогенетикой». bioRxiv 10.1101/2020.10.29.361360 .
- ^ Фиораванти Д., Джарратано И., Маджио В., Агостинелли С., Кьеричи М., Юрман Г., Фурланелло С. (март 2018 г.). «Филогенетические сверточные нейронные сети в метагеномике» . БМК Биоинформатика . 19 (Приложение 2): 49. doi : 10.1186/s12859-018-2033-5 . ПМЦ 5850953 . ПМИД 29536822 .
- ^ Дхунгел Э., Мрейуд Ю., Гвак Х.Дж., Раджех А., Ро М., Ан Т.Х. (январь 2021 г.). «MegaR: интерактивный пакет R для быстрой классификации образцов и прогнозирования фенотипов с использованием профилей метагенома и машинного обучения» . БМК Биоинформатика . 22 (1): 25. дои : 10.1186/s12859-020-03933-4 . ПМЦ 7814621 . ПМИД 33461494 .
- ^ Сюнь В., Лю Ю., Ли В., Жэнь Ю., Сюн В., Сюй З. и др. (январь 2021 г.). «Специализированные метаболические функции ключевых таксонов поддерживают стабильность микробиома почвы» . Микробиом . 9 (1): 35. дои : 10.1186/s40168-020-00985-9 . ПМЦ 7849160 . ПМИД 33517892 .
- ↑ Перейти обратно: Перейти обратно: а б Пироозния М., Ян Цзюй, Ян MQ, Дэн Юй (2008). «Сравнительное исследование различных методов машинного обучения на данных об экспрессии генов на микрочипах» . БМК Геномика . 9 Приложение 1 (1): S13. дои : 10.1186/1471-2164-9-S1-S13 . ПМК 2386055 . ПМИД 18366602 .
- ^ «Машинное обучение в молекулярной системной биологии» . Границы . Проверено 9 июня 2017 г.
- ^ д'Альше-Бюк Ф., Вехенкель Л. (декабрь 2008 г.). «Машинное обучение в системной биологии» . Дело БМК . 2 Приложение 4 (4): S1. дои : 10.1186/1753-6561-2-S4-S1 . ПМЦ 2654969 . ПМИД 19091048 .
- ^ Бхаттачарья М (2020). «Неконтролируемые методы в геномике». Ин Шриниваса М.Г., Сиддеш Г.М., Манисекхар С.Р. (ред.). Принципы статистического моделирования и машинного обучения для методов, инструментов и приложений биоинформатики . Спрингер Сингапур. стр. 164–188. ISBN 978-981-15-2445-5 .
- ^ Тополь Е.Ю. (январь 2019). «Высокоэффективная медицина: конвергенция человеческого и искусственного интеллекта». Природная медицина . 25 (1): 44–56. дои : 10.1038/s41591-018-0300-7 . ПМИД 30617339 . S2CID 57574615 .
- ↑ Перейти обратно: Перейти обратно: а б Цзян Ф., Цзян Ю., Чжи Х., Донг Ю., Ли Х., Ма С. и др. (декабрь 2017 г.). «Искусственный интеллект в здравоохранении: прошлое, настоящее и будущее» . Инсульт и сосудистая неврология . 2 (4): 230–243. дои : 10.1136/svn-2017-000101 . ПМЦ 5829945 . ПМИД 29507784 .
- ^ Мирцхулава Л., Вонг Дж., Аль-Маджид С., Пирс Дж. (март 2015 г.). «Модель искусственной нейронной сети в диагностике инсульта» (PDF) . 2015 17-я Международная конференция UKSim-AMSS по моделированию и симуляции (UKSim) . стр. 50–53. дои : 10.1109/UKSim.2015.33 . ISBN 978-1-4799-8713-9 . S2CID 6391733 .
- ^ Титано Дж.Дж., Бэджли М., Шеффляйн Дж., Пейн М., Су А., Кай М. и др. (сентябрь 2018 г.). «Автоматическое наблюдение черепных изображений с помощью глубоких нейронных сетей на предмет острых неврологических событий». Природная медицина . 24 (9): 1337–1341. дои : 10.1038/s41591-018-0147-y . ПМИД 30104767 . S2CID 51976344 .
- ↑ Перейти обратно: Перейти обратно: а б с Краллингер М., Эрхардт Р.А., Валенсия А (март 2005 г.). «Подходы к анализу текста в молекулярной биологии и биомедицине». Открытие наркотиков сегодня . 10 (6): 439–45. дои : 10.1016/S1359-6446(05)03376-3 . ПМИД 15808823 .
- ^ Пратас Д., Хоссейни М., Силва Р., Пиньо А., Феррейра П. (20–23 июня 2017 г.). «Визуализация различных участков ДНК современного человека относительно генома неандертальца». Распознавание образов и анализ изображений . Конспекты лекций по информатике. Том. 10255. стр. 235–242. дои : 10.1007/978-3-319-58838-4_26 . ISBN 978-3-319-58837-7 .
- ^ Барджетт Р.Д., Карузо Т. (март 2020 г.). «Реакция почвенного микробного сообщества на экстремальные климатические явления: устойчивость, устойчивость и переходы в альтернативные состояния» . Философские труды Лондонского королевского общества. Серия Б, Биологические науки . 375 (1794): 20190112. doi : 10.1098/rstb.2019.0112 . ПМК 7017770 . ПМИД 31983338 .
- ^ Дево А., Бонито Г., Юлинг Дж., Паолетти М., Беккер М., Биндшедлер С. и др. (май 2018 г.). «Бактериально-грибковые взаимодействия: экология, механизмы и проблемы» . Обзоры микробиологии FEMS . 42 (3): 335–352. дои : 10.1093/femsre/fuy008 . hdl : 21.11116/0000-0002-C1E7-F . ПМИД 29471481 .
- ^ Ансари М.З., Ядав Г., Гохале Р.С., Моханти Д. (июль 2004 г.). «NRPS-PKS: основанный на знаниях ресурс для анализа мегасинтаз NRPS/PKS» . Исследования нуклеиновых кислот . 32 (проблема с веб-сервером): W405-13. дои : 10.1093/nar/gkh359 . ПМК 441497 . ПМИД 15215420 .
- ↑ Перейти обратно: Перейти обратно: а б Наварро-Муньос Х.К., Селем-Мохика Н., Маллоуни М.В., Каутсар С.А., Трайон Дж.Х., Паркинсон Э.И. и др. (январь 2020 г.). «Вычислительная система для изучения крупномасштабного биосинтетического разнообразия» . Химическая биология природы . 16 (1): 60–68. дои : 10.1038/s41589-019-0400-9 . ПМЦ 6917865 . ПМИД 31768033 .
- ↑ Перейти обратно: Перейти обратно: а б Дорогази Дж.Р., Олбрайт Дж.К., Геринг А.В., Джу К.С., Хейнс Р.Р., Чалуков К.А. и др. (ноябрь 2014 г.). «Дорожная карта открытия натуральных продуктов на основе крупномасштабной геномики и метаболомики» . Химическая биология природы . 10 (11): 963–8. дои : 10.1038/nchembio.1659 . ПМК 4201863 . ПМИД 25262415 .
- ↑ Перейти обратно: Перейти обратно: а б Чимерманчик П., Медема М.Х., Клаесен Дж., Курита К., Виланд Браун Л.С., Мавромматис К. и др. (июль 2014 г.). «Понимание вторичного метаболизма на основе глобального анализа кластеров биосинтетических генов прокариот» . Клетка . 158 (2): 412–421. дои : 10.1016/j.cell.2014.06.034 . ПМК 4123684 . ПМИД 25036635 .
- ↑ Перейти обратно: Перейти обратно: а б Геринг А.В., МакКлюр Р.А., Дорогази Дж.Р., Олбрайт Дж.К., Хаверланд Н.А., Чжан Ю. и др. (февраль 2016 г.). «Метабологеномика: корреляция кластеров микробных генов с метаболитами способствует открытию нерибосомального пептида с необычным мономером аминокислоты» . Центральная научная служба ACS . 2 (2): 99–108. дои : 10.1021/accentsci.5b00331 . ПМЦ 4827660 . ПМИД 27163034 .
- ^ Амири Могаддам Дж., Круземанн М., Аланджари М., Хармс Х., Давила-Сеспедес А., Блом Дж. и др. (ноябрь 2018 г.). «Анализ генома и метаболома морских миксобактерий выявил высокий потенциал биосинтеза новых специализированных метаболитов» . Научные отчеты . 8 (1): 16600. Бибкод : 2018NatSR...816600A . дои : 10.1038/s41598-018-34954-y . ПМК 6226438 . ПМИД 30413766 .
- ^ Дункан К.Р., Круземанн М., Лехнер А., Саркар А., Ли Дж., Цимерт Н. и др. (апрель 2015 г.). «Молекулярные сети и анализ генома на основе шаблонов улучшают обнаружение кластеров биосинтетических генов и их продуктов из видов Salinispora» . Химия и биология . 22 (4): 460–471. doi : 10.1016/j.chembiol.2015.03.010 . ПМК 4409930 . ПМИД 25865308 .
- ^ Нильсен Дж.К., Грийсилс С., Приджент С., Джи Б., Дайнат Дж., Нильсен К.Ф. и др. (апрель 2017 г.). «Глобальный анализ кластеров биосинтетических генов обнаруживает огромный потенциал производства вторичных метаболитов у видов Penicillium». Природная микробиология . 2 (6): 17044. doi : 10.1038/nmicrobiol.2017.44 . ПМИД 28368369 . S2CID 22699928 .
- ^ МакКлюр Р.А., Геринг А.В., Джу К.С., Баччиле Дж.А., Шредер Ф.К., Меткалф В.В. и др. (декабрь 2016 г.). «Выяснение семейств натуральных продуктов римозамид-детоксин и их биосинтеза с использованием корреляции кластеров метаболитов и генов» . АКС Химическая биология . 11 (12): 3452–3460. дои : 10.1021/acschembio.6b00779 . ПМЦ 5295535 . ПМИД 27809474 .
- ^ Цао Л., Щербин Э., Мохимани Х. (август 2019 г.). «Сеть ассоциаций всего метаболома и метагенома выявляет микробные натуральные продукты и продукты микробной биотрансформации из микробиоты человека» . mSystems . 4 (4): e00387–19, /msystems/4/4/msys.00387–19.atom. дои : 10.1128/mSystems.00387-19 . ПМК 6712304 . ПМИД 31455639 .
- ^ Олм М.Р., Бхаттачарья Н., Критс-Кристоф А., Фирек Б.А., Бейкер Р., Сонг Ю.С. и др. (декабрь 2019 г.). «Некротизирующему энтероколиту предшествует повышенная репликация кишечных бактерий, клебсиелл и бактерий, кодирующих фимбрии» . Достижения науки . 5 (12): eaax5727. Бибкод : 2019SciA....5.5727O . дои : 10.1126/sciadv.aax5727 . ПМК 6905865 . ПМИД 31844663 .
- ^ Каррион В.Дж., Перес-Харамильо Дж., Кордовес В., Траканна В., де Холландер М., Руис-Бак Д. и др. (ноябрь 2019 г.). «Патоген-индуцированная активация функций подавления заболеваний в эндофитном корневом микробиоме» . Наука . 366 (6465): 606–612. Бибкод : 2019Sci...366..606C . дои : 10.1126/science.aaw9285 . hdl : 1887/3188901 . ПМИД 31672892 . S2CID 207814746 .
- ^ Паскаль Андреу, Виктория; Августин, Ханна Э.; ван ден Берг, Коэн; ван дер Хоофт, Джастин Джей-Джей; Фишбах, Майкл А.; Медема, Марникс Х. (26 октября 2021 г.). Шанк, Элизабет Энн (ред.). «BiG-MAP: автоматизированный конвейер для анализа численности и экспрессии кластеров метаболических генов в микробиомах» . mSystems . 6 (5): e0093721. doi : 10.1128/mSystems.00937-21 . ISSN 2379-5077 . ПМЦ 8547482 . ПМИД 34581602 .
- ^ Агравал П., Хатер С., Гупта М., Сайн Н., Моханти Д. (июль 2017 г.). «RiPPMiner: биоинформатический ресурс для расшифровки химической структуры RiPP на основе прогнозирования расщепления и поперечных связей» . Исследования нуклеиновых кислот . 45 (П1): W80–W88. дои : 10.1093/nar/gkx408 . ПМК 5570163 . ПМИД 28499008 .
- ^ Хубер Ф., Риддер Л., Верховен С., Спаакс Дж. Х., Диблен Ф., Роджерс С., ван дер Хоофт Дж. Дж. (февраль 2021 г.). «Spec2Vec: Улучшена оценка масс-спектрального сходства за счет изучения структурных взаимосвязей» . PLOS Вычислительная биология . 17 (2): e1008724. Бибкод : 2021PLSCB..17E8724H . дои : 10.1371/journal.pcbi.1008724 . ПМК 7909622 . ПМИД 33591968 .
- ^ Хубер Ф., Риддер Л., Верховен С., Спаакс Дж. Х., Диблен Ф., Роджерс С., ван дер Хоофт Дж. Дж. (февраль 2021 г.). «Spec2Vec: Улучшена оценка масс-спектрального сходства за счет изучения структурных взаимосвязей» . PLOS Вычислительная биология . 17 (2): e1008724. Бибкод : 2021PLSCB..17E8724H . дои : 10.1371/journal.pcbi.1008724 . ПМК 7909622 . ПМИД 33591968 .
- ^ Национальный центр биотехнологической информации; Национальная медицинская библиотека США. «Национальный центр биотехнологической информации» . ncbi.nlm.nih.gov . Проверено 30 июля 2021 г.
- ^ Агарвала Р., Барретт Т., Бек Дж., Бенсон Д.А., Боллин С., Болтон Е. и др. (Координаторы ресурсов NCBI) (январь 2018 г.). «Ресурсы базы данных Национального центра биотехнологической информации» . Исследования нуклеиновых кислот . 46 (Д1): Д8–Д13. дои : 10.1093/нар/gkx1095 . ПМЦ 5753372 . ПМИД 29140470 .
- ^ «база данных антиСМАШ» . antismash-db.вторичныеметаболиты.org .
- ^ «МИБиГ: Минимальная информация о кластере биосинтетических генов» . mibig.вторичныеметаболиты.org . Проверено 30 июля 2021 г.
- ^ МиБиГ
- ^ Каутсар С.А., Блин К., Шоу С., Наварро-Муньос Х.К., Терлоу Б.Р., ван дер Хоофт Дж.Дж. и др. (январь 2020 г.). «MIBiG 2.0: хранилище кластеров биосинтетических генов с известной функцией» . Исследования нуклеиновых кислот . 48 (Д1): Д454–Д458. дои : 10.1093/nar/gkz882 . ПМЦ 7145714 . ПМИД 31612915 .
- ^ «Сильва» . arb-silva.de . Проверено 30 июля 2021 г.
- ^ Кваст С., Прюссе Е., Йилмаз П., Геркен Дж., Швеер Т., Ярза П. и др. (январь 2013 г.). «Проект базы данных генов рибосомальных РНК SILVA: улучшенная обработка данных и веб-инструменты» . Исследования нуклеиновых кислот . 41 (Проблема с базой данных): D590-6. дои : 10.1093/nar/gks1219 . ПМЦ 3531112 . ПМИД 23193283 .
- ^ «greengenes. Secondgenome.com» . greengenes . Secondgenome.com . Проверено 30 июля 2021 г.
- ^ ДеСантис Т.З., Хугенхольц П., Ларсен Н., Рохас М., Броди Э.Л., Келлер К. и др. (июль 2006 г.). «Greengenes, проверенная химерами база данных генов 16S рРНК и инструментальные средства, совместимые с ARB» . Прикладная и экологическая микробиология . 72 (7): 5069–72. Бибкод : 2006ApEnM..72.5069D . дои : 10.1128/АЕМ.03006-05 . ПМЦ 1489311 . ПМИД 16820507 .
- ^ Макдональд Д., Прайс М.Н., Гудрич Дж., Навроцкий Е.П., ДеСантис Т.З., Пробст А. и др. (март 2012 г.). «Улучшенная таксономия Greengenes с четкими рангами для экологического и эволюционного анализа бактерий и архей» . Журнал ISME . 6 (3): 610–8. Бибкод : 2012ISMEJ...6..610M . дои : 10.1038/ismej.2011.139 . ПМК 3280142 . ПМИД 22134646 .
- ^ «открытое дерево» . Tree.opentreeoflife.org . Проверено 30 июля 2021 г.
- ^ Хинчлифф С.Э., Смит С.А., Оллман Дж.Ф., Берли Дж.Г., Чаудхари Р., Когхилл Л.М. и др. (октябрь 2015 г.). «Синтез филогении и таксономии в комплексное древо жизни» . Труды Национальной академии наук Соединенных Штатов Америки . 112 (41): 12764–9. Бибкод : 2015PNAS..11212764H . дои : 10.1073/pnas.1423041112 . ПМЦ 4611642 . ПМИД 26385966 .
- ^ «Выпуск 11 RDP — инструменты анализа последовательностей» . rdp.cme.msu.edu . Архивировано из оригинала 19 августа 2020 года . Проверено 30 июля 2021 г.
- ^ Коул-младший, Ван К., Фиш Дж.А., Чай Б., МакГаррелл Д.М., Сунь Ю. и др. (январь 2014 г.). «Проект рибосомальной базы данных: данные и инструменты для высокопроизводительного анализа рРНК» . Исследования нуклеиновых кислот . 42 (Проблема с базой данных): D633-42. дои : 10.1093/nar/gkt1244 . ПМЦ 3965039 . ПМИД 24288368 .
Дифференцируемые вычисления |
|---|