Машинное обучение в биоинформатике
| Часть серии на |
| Искусственный интеллект |
|---|
Машинное обучение в биоинформатике - это применение алгоритмов машинного обучения к биоинформатике , [ 1 ] включая геномику , протеомику , микрочипы , системную биологию , эволюцию и добычу текста . [ 2 ] [ 3 ]
До появления машинного обучения алгоритмы биоинформатики должны были запрограммироваться вручную; Для таких проблем, как прогноз структуры белка , это оказалось трудным. [ 4 ] Методы машинного обучения, такие как глубокое обучение, могут изучать функции наборов данных, а не требовать от программиста определять их индивидуально. Алгоритм может дополнительно научиться объединять функции низкого уровня в более абстрактные особенности и так далее. Этот многослойный подход позволяет таким системам делать сложные прогнозы при подходящем обучении. Эти методы контрастируют с другими подходами к вычислительной биологии, которые, одновременно эксплуатируя существующие наборы данных, не позволяют интерпретировать и анализировать данные непредвиденными способами.
Задачи
[ редактировать ]Алгоритмы машинного обучения в биоинформатике могут использоваться для прогнозирования, классификации и выбора функций. Методы для достижения этой задачи разнообразны и охватывают многие дисциплины; Наиболее известными среди них являются машинное обучение и статистика. Задачи классификации и прогнозирования направлены на создание моделей, которые описывают и различают классы или концепции для будущего прогнозирования. Различия между ними следующие:
- Классификация/распознавание выводит категорический класс, в то время как прогноз выводит численную ценную функцию.
- Тип алгоритма или процесса, используемый для создания прогнозирующих моделей из данных с использованием аналогий, правил, нейронных сетей, вероятностей и/или статистики.
В связи с экспоненциальным ростом информационных технологий и применимых моделей, включая искусственный интеллект и интеллектуальное анализ данных, в дополнение к вечно более подробным наборам данных доступа, были созданы новые и лучшие методы анализа информации, основанные на их способности учиться. Такие модели допускают охват за пределами описания и предоставляют понимание в виде тестируемых моделей.
Подходы машинного обучения
[ редактировать ]Искусственные нейронные сети
[ редактировать ]Искусственные нейронные сети в биоинформатике использовались для: [ 5 ]
- Сравнение и выравнивание последовательностей РНК, белка и ДНК.
- Идентификация промоторов и поиск генов из последовательностей, связанных с ДНК.
- Интерпретация данных выражения и микроаррея.
- Выявление сети (регулирующих) генов.
- Изучение эволюционных отношений путем построения филогенетических деревьев .
- Классификация и прогнозирование структуры белка .
- Молекулярный дизайн и стыковка
Функциональная инженерия
[ редактировать ]То, как функции, часто векторы в многомерном пространстве, извлекаются из данных домена, является важным компонентом систем обучения. [ 6 ] В геномике типичным представлением последовательности является вектор частот K-MERS , который является вектором измерения чьи записи учитывают появление каждой последующей длины в данной последовательности. Поскольку для столь же значения, как Размерность этих векторов огромна (например, в этом случае измерение ), такие методы, как анализ основных компонентов, используются для проецирования данных в более низкое пространство, тем самым выбирая меньший набор функций из последовательностей. [ 6 ] [ Дополнительные цитаты (ы) необходимы ]




Классификация
[ редактировать ]В этом типе задачи машинного обучения вывод является дискретной переменной. Одним из примеров этого типа задачи в биоинформатике является маркировка новых геномных данных (таких как геномы некультурируемых бактерий) на основе модели уже помеченных данных. [ 6 ]
Скрытые модели Маркова
[ редактировать ]Скрытые модели Маркова (HMM) представляют собой класс статистических моделей для последовательных данных (часто связанные с системами, развивающимися с течением времени). HMM состоит из двух математических объектов: наблюдаемый зависимый от состояния процесс и ненаблюдаемый (скрытый) государственный процесс Полем В HMM процесс состояния не наблюдается напрямую - это «скрытая» (или «скрытая») переменная - но наблюдения сделаны из зависимого от состояния процесса (или процесса наблюдения), который обусловлен основным процессом состояния ( и который, таким образом, можно рассматривать как шумное измерение интересующих состояний системы). [ 7 ] HMM могут быть сформулированы в непрерывное время. [ 8 ] [ 9 ]


HMMS можно использовать для профилирования и преобразования выравнивания множественных последовательностей в систему оценки специфики для положения, подходящую для поиска баз данных для гомологичных последовательностей удаленно. [ 10 ] Кроме того, экологические явления могут быть описаны HMMS. [ 11 ]
Сверточные нейронные сети
[ редактировать ]Свещательные нейронные сети (CNN) представляют собой класс глубокой нейронной сети , архитектура которых основана на общих весах ядер или фильтров свертки, которые скользят по входным характеристикам, обеспечивая ответы-перевод, известные как карты признаков. [ 12 ] [ 13 ] CNN используют преимущества иерархической паттерны в данных и собирают паттерны увеличивающейся сложности, используя меньшие и более простые паттерны, обнаруженные через их фильтры. [ 14 ]
Сверточные сети вдохновлены биологическими процессами были [ 15 ] [ 16 ] [ 17 ] [ 18 ] В том, что схема связности между нейронами напоминает организацию зрительной коры животных . Индивидуальные корковые нейроны реагируют на стимулы только в ограниченной области визуального поля, известной как восприимчивое поле . Восприимчивые поля разных нейронов частично перекрываются так, чтобы они покрывали все поле зрения.
CNN использует относительно небольшую предварительную обработку по сравнению с другими алгоритмами классификации изображений . Это означает, что сеть учится оптимизировать фильтры (или ядра) с помощью автоматического обучения, тогда как в традиционных алгоритмах эти фильтры вручную . Эта сниженная зависимость от предварительного знания аналитика и вмешательства человека в ручную извлечение объектов делает CNNs желательной моделью. [ 14 ]
Филогенетическая сверточная нейронная сеть (PH-CNN) представляет собой сверточную архитектуру нейронной сети, предложенную Fioranti et al. В 2018 году классифицировать данные метагеномики . [ 19 ] При таком подходе филогенетические данные наделены патристическим расстоянием (сумма длины всех ветвей, соединяющих два операционных таксономических единиц [OTU]), чтобы выбрать K-neighbore для каждого OTU, и каждый OTU и его соседи обрабатываются в коннусоотешенные фильтры.
Самоотверженное обучение
[ редактировать ]В отличие от контролируемых методов, самоотверженные методы обучения изучают представления, не полагаясь на аннотированные данные. Это хорошо подходит для геномики, где методы высокопроизводительной секвенирования могут создавать потенциально большие объемы немеченых данных. Некоторые примеры самоподдерживаемых методов обучения, применяемых на геномике, включают DNABERT и Self-Genomenet. [ 20 ] [ 21 ]
Случайный лес
[ редактировать ]
Случайные леса (RF) классифицируют путем построения ансамбля деревьев решений и выводя средний прогноз отдельных деревьев. [ 22 ] Это модификация агрегирования начальной загрузки (которая объединяет большую коллекцию деревьев решений) и может использоваться для классификации или регрессии . [ 23 ] [ 24 ]
Поскольку случайные леса дают внутреннюю оценку ошибки обобщения, перекрестная проверка является ненужной. Кроме того, они производят близость, которая может использоваться для вменения пропущенных значений, и которые позволяют обеспечить новые визуализации данных. [ 25 ]
Вычислительно, случайные леса привлекательны, потому что они естественным образом обрабатывают как регрессию, так и (мультиклассную) классификацию, относительно быстры для обучения и прогнозирования, зависят только от одного или двух параметров настройки, могут быть использованы встроенной оценкой ошибки обобщения, можно использовать. непосредственно для высокомерных проблем и может быть легко реализована параллельно. Статистически случайные леса привлекательны для дополнительных функций, таких как показатели переменной важности, дифференциальное взвешивание класса, вменение недостающей стоимостью, визуализация, обнаружение выбросов и неконтролируемое обучение. [ 25 ]
Кластеризация
[ редактировать ]Кластеризация - разделение данных, установленных на непрерывные подмножества, так что данные в каждом подмножестве были максимально близки друг к другу и как можно дальше от данных в любом другом подмножестве, в соответствии с некоторой определенной функцией расстояния или сходства - является Общий метод для статистического анализа данных.
Кластеризация является центральной для многих исследований биоинформатики, основанных на данных и служит мощным вычислительным методом, посредством которого средства иерархической, на основе центроида, на основе распределения, на основе плотности и самоорганизации классификации карт давно изучались и используются в классической машине Учебные настройки. В частности, кластеризация помогает анализировать неструктурированные и высокомерные данные в виде последовательностей, выражений, текстов, изображений и так далее. Кластеризация также используется для получения понимания биологических процессов на геномном уровне, например, функциях генов, клеточных процессах, подтипах клеток, регуляции генов и метаболических процессах. [ 26 ]
Алгоритмы кластеризации, используемые в биоинформатике
[ редактировать ]Алгоритмы кластеризации данных могут быть иерархическими или частичными. Иерархические алгоритмы находят последовательные кластеры с использованием ранее установленных кластеров, тогда как разделы, а алгоритмы определяют все кластеры одновременно. Иерархические алгоритмы могут быть агломеративными (снизу вверх) или разделительными (сверху вниз).
Агломеративные алгоритмы начинаются с каждого элемента как отдельного кластера и объединяют их в последовательно больших кластерах. Разделительные алгоритмы начинаются со всего набора и продолжают разделить его на последовательно меньшие кластеры. Иерархическая кластеризация рассчитывается с использованием метрик на евклидовых пространствах , наиболее часто используемым является евклидовое расстояние, рассчитанное путем поиска квадрата разницы между каждой переменной, добавляя все квадраты и обнаружив квадратный корень указанной суммы. Примером иерархического алгоритма кластеризации является Береза , которая особенно хороша в биоинформатике в течение ее почти линейной временной сложности, с учетом в целом больших наборов данных. [ 27 ] Алгоритмы разделения основаны на определении начального количества групп и итеративно перераспределяющим объектами между группами для сходимости. Этот алгоритм обычно определяет все кластеры одновременно. Большинство приложений принимают один из двух популярных эвристических методов: алгоритм K-средних или K-медиаиды . Другие алгоритмы не требуют начального количества групп, таких как распространение аффинности . В геномной обстановке этот алгоритм использовался как для кластера биосинтетических кластеров генов в семействах кластеров генов (GCF), так и для кластера указанных GCF. [ 28 ]
Рабочий процесс
[ редактировать ]Как правило, рабочий процесс для применения машинного обучения к биологическим данным проходит через четыре шага: [ 2 ]
- Запись, включая захват и хранение. На этом этапе различные источники информации могут быть объединены в один набор.
- Предварительная обработка, включая очистку и реструктуризацию в готовую к анализу. На этом этапе некорректированные данные устраняются или исправлены, в то время как отсутствующие данные могут вменены и выбранные соответствующие переменные.
- Анализ, оценка данных с использованием контролируемых или неконтролируемых алгоритмов. Алгоритм обычно обучается на подмножестве данных, оптимизируется параметры и оценивается на отдельном подмножестве теста.
- Визуализация и интерпретация, где знание представлено эффективно с использованием различных методов для оценки значимости и важности результатов.
Ошибки данных
[ редактировать ]- Дубликаты данных являются важной проблемой в биоинформатике. Общедоступные данные могут быть неопределенного качества. [ 29 ]
- Ошибки во время экспериментов. [ 29 ]
- Ошибочная интерпретация. [ 29 ]
- Печатать ошибки. [ 29 ]
- Нестандартизированные методы (3D-структура в PDB из нескольких источников, рентгеновская дифракция, теоретическое моделирование, ядерный магнитный резонанс и т. Д.) Используются в экспериментах. [ 29 ]
Приложения
[ редактировать ]В целом, систему машинного обучения обычно может быть обучена распознавать элементы определенного класса с учетом достаточного количества образцов. [ 30 ] Например, методы машинного обучения могут быть обучены для определения конкретных визуальных функций, таких как сайты сплайсинга. [ 31 ]
Векторные машины поддержки широко использовались в исследованиях генома рака. [ 32 ] Кроме того, глубокое обучение было включено в биоинформационные алгоритмы. Приложения глубокого обучения использовались для регуляторной геномики и клеточной визуализации. [ 33 ] Другие приложения включают в себя классификацию медицинских изображений, анализ геномных последовательностей, а также классификацию и прогноз структуры белка. [ 34 ] Глубокое обучение было применено к регуляторной геномике, вариантам вызова и оценки патогенности. [ 35 ] Обработка естественного языка и добыча текста помогли понять явления, включая взаимодействие белкового белка, отношение генов-дизаза, а также прогнозирование биомолекулярных структур и функций. [ 36 ]
Точность/персонализированная медицина
[ редактировать ]обработки естественного языка Алгоритмы Персонализированная медицина для пациентов, страдающих генетическими заболеваниями, путем объединения экстракции клинической информации и геномных данных, доступных у пациентов. Такие институты, как сеть исследований в области фармакогеномики, финансируемая здоровьем, фокусируется на поиске лечения рака молочной железы. [ 37 ]
Прецизионная медицина рассматривает индивидуальную геномную изменчивость, обеспечиваемой крупномасштабными биологическими базами данных. Машинное обучение может быть применено для выполнения функции соответствия между (группами пациентов) и конкретными методами лечения. [ 38 ]
Вычислительные методы используются для решения других проблем, таких как эффективная конструкция праймеров для ПЦР , анализ биологического изображения и трансляция белков на спине (то есть, учитывая дегенерацию генетического кода, сложная комбинаторная задача). [ 2 ]
Геномика
[ редактировать ]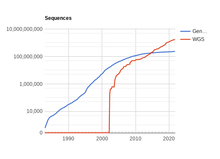
Хотя данные о геномной последовательности исторически были редкими из -за технической сложности секвенирования куски ДНК, количество доступных последовательностей растет. В среднем количество баз , доступных в общественном хранилище Genbank, удвоилось каждые 18 месяцев с 1982 года. [ 39 ] Однако, хотя необработанные данные становятся все более доступными и доступными, по состоянию на 2002 год [update]Биологическая интерпретация этих данных происходила гораздо более медленными темпами. [ 40 ] Это создано для растущей потребности в разработке инструментов вычислительной геномики , включая системы машинного обучения, которые могут автоматически определять местоположение генов, кодирующих белок, в данной последовательности ДНК (IE прогнозирование генов ). [ 40 ]
Предсказание генов обычно выполняется как с помощью внешних поисков , так и с внутренним поиском . [ 40 ] Для внешнего поиска последовательность входной ДНК проводится через большую базу данных последовательностей, чьи гены были ранее обнаружены, а их местоположения аннотируют и идентифицирующие гены целевой последовательности, определяя, какие строки оснований в последовательности гомологичны известной генной последовательностям. Однако не все гены в данной входной последовательности могут быть идентифицированы только с помощью гомологии из -за ограничений в размере базы данных известных и аннотированных последовательностей генов. Следовательно, необходим внутренний поиск, когда программа прогнозирования генов пытается идентифицировать оставшиеся гены только от последовательности ДНК. [ 40 ]
Машинное обучение также использовалось для проблемы выравнивания множественных последовательностей , которая включает в себя выравнивание многих последовательностей ДНК или аминокислот, чтобы определить области сходства, которые могут указывать на общую историю эволюции. [ 2 ] Его также можно использовать для обнаружения и визуализации перестроек генома. [ 41 ]
Протеомика
[ редактировать ]
Белки , струны аминокислот , получают большую часть своей функции от складывания белков , где они соответствуют трехмерной структуре, включая первичную структуру , вторичную структуру ( альфа-спирали и бета-листы ), третичная структура и четвертичная структура Полем
Прогнозирование вторичной структуры белка является основным направлением этого подпола, поскольку третичные и четвертичные структуры определяются на основе вторичной структуры. [ 4 ] Решение истинной структуры белка является дорогостоящим и интенсивным по времени, что способствует необходимости в системах, которые могут точно предсказать структуру белка, путем непосредственного анализа аминокислотной последовательности. [ 4 ] [ 2 ] До машинного обучения исследователи должны были провести этот прогноз вручную. Эта тенденция началась в 1951 году, когда Полинг и Кори опубликовали свою работу по прогнозированию конфигураций водородных связей белка из полипептидной цепи. [ 42 ] Автоматическое обучение функций достигает точности 82-84%. [ 4 ] [ 43 ] В текущем современном прогнозе вторичной структуры используется система, называемая DeepCNF (глубокие сверточные нейронные поля), которая опирается на модель машинного обучения искусственных нейронных сетей для достижения точности приблизительно 84%, когда ему поручено классифицировать аминокислоты белковой последовательности в один из трех структурных классов (спираль, лист или катушка). [ 43 ] [ нуждается в обновлении ] Теоретический предел для вторичной структуры белка с тремя государствами составляет 88–90%. [ 4 ]
Машинное обучение также применяется к протеомическим проблемам, таким как прогноз боковой цепи белки , моделирование белковой петли и прогноз контакта белка . [ 2 ]
Метагеномика
[ редактировать ]Метагеномика - это изучение микробных сообществ из образцов ДНК окружающей среды. [ 44 ] В настоящее время ограничения и проблемы преобладают в реализации инструментов машинного обучения из -за объема данных в образцах окружающей среды. [ 45 ] Суперкомпьютеры и веб -серверы облегчили доступ к этим инструментам. [ 46 ] Высокая размерность наборов данных микробиомов является серьезной проблемой при изучении микробиома; Это значительно ограничивает силу текущих подходов для выявления истинных различий и увеличивает вероятность ложных открытий. [ 47 ] [ Лучший источник необходим ]
были сосредоточены на изучении кишечной микробиоты и взаимосвязи с пищеварительными заболеваниями, такими как воспалительные заболевания кишечника (IBD), клостридиоиды инфекции (CDI), рак колоректального связанные с метагеномикой , Несмотря на их значение, инструменты машинного обучения , лечение. [ 46 ] Многие алгоритмы были разработаны для классификации микробных сообществ в соответствии с состоянием здоровья хозяина, независимо от типа данных о последовательности, например, rRNA 16S или секвенирования всего генома (WGS), используя такие методы, как наименьшее абсолютное усадку и классификатор оператора отбора, случайный Лес , контролируемая классификационная модель и модель дерева повышенной градиента. нейронные сети , такие как рецидивирующие нейронные сети (RNN), сверточные нейронные сети (CNN) и нейронные сети Хопфилда . Были добавлены [ 46 ] Например, в 2018 году Fioravanti et al. Разработал алгоритм, называемый pH-CNN для классификации образцов данных от здоровых пациентов и пациентов с симптомами ВЗК (для различения здоровых и больных пациентов), используя филогенетические деревья и сверточные нейронные сети. [ 48 ]
Кроме того, методы случайных лесов (RF) и внедренные показатели важности помогают в идентификации видов микробиомов, которые можно использовать для отличия больных и неосвязных образцов. Тем не менее, эффективность дерева решений и разнообразие деревьев решений в ансамбле значительно влияет на производительность радиочастотных алгоритмов. Ошибка обобщения для РФ измеряет, насколько точны отдельные классификаторы и их взаимозависимость. Следовательно, проблемы с высокой размерностью наборов данных микробиома создают проблемы. Эффективные подходы требуют много возможных переменных комбинаций, которые экспоненциально увеличивают вычислительную нагрузку по мере увеличения количества функций. [ 47 ]
Для анализа микробиомов в 2020 году Dang & Kishino [ 47 ] Разработал новый аналитический конвейер. Ядро трубопровода представляет собой радиочастотный классификатор, в сочетании с выбором переменной пересылки (RF-FVS), который выбирает набор ядра минимального размера микробных видов или функциональных сигнатур, которые максимизируют производительность прогнозирующего классификатора. Структура объединяется:
- Определение нескольких важных функций с помощью массовой параллельной выбора переменной форварда процедуры
- Картирование выбранных видов на филогенетическом дереве и
- Прогнозирование функциональных профилей с помощью функционального анализа обогащения генов из метагеномных данных 16S рРНК .
Они продемонстрировали производительность, анализируя два опубликованных набора данных из крупномасштабных исследований по случаю контроля:
- Данные ампликона гена 16S рРНК для инфекции C. difficile (CDI) и
- Данные о метагеномике с дробовиком для рака колоректального рака человека (CRC).
Предложенный подход повысил точность с 81% до 99,01% для CDI и с 75,14% до 90,17% для CRC.
Использование машинного обучения в образцах окружающей среды было менее изучено, возможно, из -за сложности данных, особенно из WGS. Некоторые работы показывают, что можно применить эти инструменты в образцах окружающей среды. В 2021 году Dhungel et al., [ 49 ] спроектировал пакет R под названием Megar. Этот пакет позволяет работать с 16S рРНК и целыми метагеномными последовательностями, чтобы сделать таксономические профили и модели классификации с помощью моделей машинного обучения. Мегар включает в себя удобную среду визуализации, чтобы улучшить пользовательский опыт. Машинное обучение в экологической метагеномике может помочь ответить на вопросы, связанные с взаимодействием между микробными сообществами и экосистемами, например, на работу Xun et al., В 2021 году. [ 50 ] где использование различных методов машинного обучения дало представление о взаимосвязи между почвой, биоразнообразием микробиома и стабильности экосистемы.
Микрочипы
[ редактировать ]Микрочипы , тип лаборатории на чипе , используются для автоматического сбора данных о больших количествах биологического материала. Машинное обучение может помочь в анализе и применяется к идентификации паттерна экспрессии, классификации и генетической сети. [ 2 ]
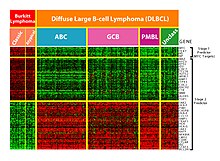
Эта технология особенно полезна для мониторинга экспрессии генов, помогая диагностировать рак, изучая, какие гены экспрессируются. [ 51 ] Одной из основных задач является определение того, какие гены экспрессируются на основе собранных данных. [ 2 ] Кроме того, из -за огромного количества генов, на которых данные собирают микрочипость, в результате чего большое количество не относящихся к делу данных задача экспрессированной идентификации генов является сложной задачей. Машинное обучение представляет потенциальное решение, так как для выполнения этой идентификации могут использоваться различные методы классификации. Наиболее часто используемыми методами являются радиальные базисные функциональные сети , глубокое обучение , байесовская классификация , деревья решений и случайный лес . [ 51 ]
Системная биология
[ редактировать ]Системная биология фокусируется на изучении возникающего поведения из сложных взаимодействий простых биологических компонентов в системе. Такие компоненты могут включать ДНК, РНК, белки и метаболиты. [ 52 ]
Машинное обучение использовалось для помощи в моделировании этих взаимодействий в таких областях, как генетические сети, сети передачи сигнала и метаболические пути. [ 2 ] Вероятностные графические модели , метод машинного обучения для определения взаимосвязи между различными переменными, являются одним из наиболее часто используемых методов для моделирования генетических сетей. [ 2 ] Кроме того, машинное обучение было применено к проблемам системной биологии, таким как идентификация сайтов связывания транскрипционного фактора с использованием оптимизации цепи Маркова . [ 2 ] Генетические алгоритмы , методы машинного обучения, которые основаны на естественном процессе эволюции, использовались для моделирования генетических сетей и регуляторных структур. [ 2 ]
Другие системы биологии системных биологии включают задачу прогнозирования функции фермента, анализ данных микрочипов с высокой пропускной способностью, анализ исследований в области ассоциаций по всему геному для лучшего понимания маркеров заболевания, прогнозирования функции белка. [ 53 ]
Эволюция
[ редактировать ]Этот домен, особенно реконструкция филогенетического дерева , использует особенности методов машинного обучения. Филогенетические деревья являются схематическими представлениями эволюции организмов. Первоначально они были построены с использованием таких особенностей, как морфологические и метаболические особенности. Позже, из -за наличия последовательностей генома, конструкция алгоритма филогенетического дерева использовала концепцию, основанную на сравнении генома. С помощью методов оптимизации сравнение было проведено с помощью множественного выравнивания последовательности. [ 54 ]
Диагноз инсульта
[ редактировать ]Методы машинного обучения для анализа данных нейровизуализации используются для диагностики инсульта . Исторически многочисленные подходы к этой проблеме включали нейронные сети. [ 55 ] [ 56 ]
Несколько подходов к обнаружению инсультов использовали машинное обучение. Как предложено Мирцхулавой, [ 57 ] Сеты подачи были протестированы для обнаружения инсультов с использованием нейронной визуализации. Как предложено Титано [ 58 ] Методы 3D-CNN были протестированы в контролируемой классификации для скрининга КТ-изображений для острых неврологических событий. Трехмерные методы CNN и SVM часто используются. [ 56 ]
Текст добыча
[ редактировать ]Увеличение биологических публикаций увеличило сложность поиска и составления соответствующей доступной информации по данной теме. Эта задача известна как извлечение знаний . Это необходимо для сбора биологических данных, которые затем, в свою очередь, могут быть включены в алгоритмы машинного обучения для получения новых биологических знаний. [ 2 ] [ 59 ] Машинное обучение может использоваться для этой задачи извлечения знаний с использованием таких методов, как обработка естественного языка, для извлечения полезной информации из отчетов, созданных человеком, в базе данных. Текст гвоздь , альтернативный подход к машинному обучению, способный извлечь особенности из клинических нот повествования, был введен в 2017 году.
Этот метод был применен для поиска новых целей лекарств, так как эта задача требует изучения информации, хранящейся в биологических базах данных и журналах. [ 59 ] Аннотации белков в базах данных белков часто не отражают полный известный набор знаний каждого белка, поэтому дополнительная информация должна быть извлечена из биомедицинской литературы. Машинное обучение было применено к автоматической аннотации функции генов и белков, определения субклеточной локализации белка , анализа массива ДНК-экспрессии , широкомасштабного анализа взаимодействия белка и анализа взаимодействия молекул. [ 59 ]
Другим применением добычи текста является обнаружение и визуализация различных областей ДНК, с которыми сталкиваются достаточные эталонные данные. [ 60 ]
Кластеризация и профилирование численности биосинтетических генов кластеров
[ редактировать ]Микробные сообщества - это сложные сборки разнообразных микроорганизмов, [ 61 ] где Symbiont Partners постоянно производит разнообразные метаболиты, полученные из первичного и вторичного (специализированного) метаболизма, из которого метаболизм играет важную роль в микробном взаимодействии. [ 62 ] Метагеномные и метаттранскриптомные данные являются важным источником для расшифровки сигналов связи.
Молекулярные механизмы производят специализированные метаболиты по -разному. Biosynthetic Gene Clusters (BGCs) attract attention, since several metabolites are clinically valuable, anti-microbial, anti-fungal, anti-parasitic, anti-tumor and immunosuppressive agents produced by the modular action of multi-enzymatic, multi-domains gene clusters, такие как нерибосомные пептид -синтетазы (NRPSS) и поликетид -синтазы (PKSS). [ 63 ] Разнообразные исследования [ 64 ] [ 65 ] [ 66 ] [ 67 ] [ 68 ] [ 69 ] [ 70 ] [ 71 ] Покажите, что группировка BGC, которые разделяют гомологичные гены основных генов в семейства генов (GCF), может дать полезную информацию о химическом разнообразии анализируемых штаммов и может поддерживать связь BGC со вторичными метаболитами. [ 65 ] [ 67 ] GCF использовались в качестве функциональных маркеров в исследованиях здоровья человека [ 72 ] [ 73 ] и изучить способность почвы подавлять грибковые патогены. [ 74 ] Учитывая их прямую связь с каталитическими ферментами и соединениями, полученными по их кодируемым путям, BGCS/GCF могут служить прокси для изучения химического пространства микробного вторичного метаболизма. Каталог GCF в секвенированных микробных геномах дает обзор существующего химического разнообразия и дает представление о будущих приоритетах. [ 64 ] [ 66 ] Такие инструменты, как большая часть и большая карта [ 75 ] появились с единственной целью раскрытия важности BGC в естественных условиях.
Декодификация химических структур Ripps
[ редактировать ]Увеличение экспериментально охарактеризованных рибосомно синтезированных и посттрансляционно модифицированных пептидов (RIPPS) вместе с наличием информации о их последовательности и химической структуре, выбранных из баз данных, таких как бублик, бактибаз, мибиг и тиобаза, предоставлена возможность разработать машину Инструменты обучения для декодирования химической структуры и классификации их.
В 2017 году исследователи из Национального института иммунологии Нью -Дели, Индия, разработали Rippminer [ 76 ] Программное обеспечение, ресурс биоинформатики для декодирования химических структур RIPP путем майнинга генома. Веб -сервер Rippminer состоит из интерфейса запроса и базы данных RIPPDB. Rippminer определяет 12 подклассов Ripps, прогнозируя сайт расщепления лидера пептида и окончательную поперечную связь химической структуры Ripp.
Массовое сходство сходства
[ редактировать ]Многие исследования метаболомики на основе тандемной масс ( MS/MS ) , такие как сопоставление библиотеки и молекулярная сеть, используют спектральное сходство в качестве прокси для структурного сходства. Spec2Vec [ 77 ] Алгоритм обеспечивает новый способ спектрального сходства, основанный на Word2VEC . Spec2VEC изучает фрагментные отношения в большом наборе спектральных данных, чтобы оценить спектральные сходства между молекулами и классифицировать неизвестные молекулы посредством этих сравнений.
Для системных аннотаций некоторые исследования метаболомики основаны на подгонке измеренных масс -спектров фрагментации к спектрам библиотеки или контрастным спектрам посредством сетевого анализа. Функции оценки используются для определения сходства между парами фрагментных спектров в рамках этих процессов. До настоящего времени ни одно исследование не предлагает результатов, которые значительно отличаются от общеиспользованного сходства на основе косинусов . [ 78 ]
Базы данных
[ редактировать ]Важной частью биоинформатики является управление большими наборами, известными как базы данных. Базы данных существуют для каждого типа биологических данных, например, для биосинтетических кластеров генов и метагеномов.
Общие базы данных по биоинформатике
[ редактировать ]Национальный центр информации о биотехнологии
[ редактировать ]Национальный центр информации о биотехнологии (NCBI) [ 79 ] Обеспечивает большой набор онлайн -ресурсов для биологической информации и данных, включая GenBank базу данных последовательности нуклеиновых кислот и базу данных PubMed, цитаты и рефераты для опубликованных журналов Life Science. Увеличение многих веб -приложений представляет собой пользовательские реализации программы BLAST, оптимизированной для поиска специализированных наборов данных. Ресурсы включают в себя управление данными PubMed, функциональные элементы Refseq, загрузку данных генома, API Variation Services, Magic-Blast, QuickBlastp и идентичные белковые группы. Все эти ресурсы можно получить через NCBI. [ 80 ]
Анализ биоинформатики для биосинтетических генов кластеров
[ редактировать ]Антисмиш
[ редактировать ]Antismash позволяет быстро идентифицировать, аннотацию и анализ средних генов биосинтеза вторичного метаболита. Он интегрирует и сшивает с большим количеством инструментов анализа вторичного метаболита в силико . [ 81 ]
Гутсмаш
[ редактировать ]Gutsmash - это инструмент, который систематически оценивает бактериальный метаболический потенциал, прогнозируя как известные, так и новые анаэробные кишечника кластеры метаболических генов (MGC) из микробиома .
Лабиг
[ редактировать ]Любовь, [ 82 ] Минимальная информация о спецификации генов биосинтетических генов предоставляет стандарт для аннотаций и метаданных на кластерах генов биосинтеза и их молекулярных продуктах. Mibig - это проект консорциума по геномным стандартам, который основан на минимальной информации о любой структуре последовательности (миксы). [ 83 ]
Мибиг облегчает стандартизированное осаждение и поиск данных биосинтетических генов, а также разработку комплексных инструментов сравнительного анализа. Он расширяет возможности следующего поколения в области биосинтеза, химии и экологии широких классов социально значимых биологически активных вторичных метаболитов , руководствуясь надежными экспериментальными данными и богатыми компонентами метаданных. [ 84 ]
Сильва
[ редактировать ]Сильва [ 85 ] это междисциплинарный проект среди биологов и компьютеров, собирающих полную базу данных РНК -рибосомальных (рРНК) последовательностей генов, как небольших ( 16S , 18S , SSU), так и крупных ( 23S , 28S , LSU) субъединиц, которые принадлежат бактериям, Archaea и домены Eukarya. Эти данные свободно доступны для академического и коммерческого использования. [ 86 ]
Зеленые
[ редактировать ]Зеленые [ 87 ] является полноразмерной базой данных гена 16S рРНК , которая обеспечивает скрининг химеры, стандартное выравнивание и кураторную таксономию на основе вывода дерева de novo. [ 88 ] [ 89 ] Обзор:
- 1 012 863 последовательности РНК из 92 684 организмов способствовали RNACENTRAL.
- Самая короткая последовательность имеет 1253 нуклеотида, самые длинные 2368.
- Средняя длина составляет 1402 нуклеотида.
- Версия базы данных: 13.5.
Таксономия открытого дерева жизни
[ редактировать ]Открытое дерево жизни таксономия (OTT) [ 90 ] Целью создания полного, динамичного и цифрового доступного дерева жизни путем синтеза опубликованных филогенетических деревьев наряду с таксономическими данными. Филогенетические деревья были классифицированы, выровнены и объединены. Таксономии были использованы для заполнения разреженных областей и зазоров, оставленных филогениями. OTT - это база, которая мало использовалась для анализа секвенирования области 16S, однако она имеет большее количество последовательностей, классифицированных таксономически до уровня рода по сравнению с Сильвой и Грингенами. Однако с точки зрения классификации на уровне края, он содержит меньшее количество информации [ 91 ]
Проект рибосомной базы данных
[ редактировать ]Проект рибосомной базы данных (RDP) [ 92 ] является базой данных, которая обеспечивает РНК -рибосомальные (рРНК) последовательности небольших субъединиц доменной бактериальной и архиальной ( 16S ); и грибные последовательности рРНК больших субъединиц ( 28S ). [ 93 ]
Ссылки
[ редактировать ]- ^ Chicco D (декабрь 2017 г.). «Десять быстрых советов для машинного обучения в вычислительной биологии» . Биоданная добыча . 10 (35): 35. doi : 10.1186/s13040-017-0155-3 . PMC 5721660 . PMID 29234465 .
- ^ Jump up to: а беременный в дюймовый и фон глин час я Дж k л м Larrañaga P, Calvo B, Santana R, Bielza C, Galdiano J, Inza I, et al. (Март 2006 г.). «Машинное обучение в биоинформатике » Брифинги в биоинформатике 7 (1): 86–1 Doi : 10.1093/ bib/ bbk0 PMID 16761367
- ^ Pérez-Wohlfeil E, Torrenoa O, Bellis LJ, Fernandes PL, Leskosek B, Trellesa O (декабрь 2018). «Обучение биоинформатиков в высокопроизводительных вычислениях» . Гелион . 4 (12): E01057. Bibcode : 2018Heliy ... 401057p . doi : 10.1016/j.heliyon.2018.e01057 . PMC 6299036 . PMID 30582061 .
- ^ Jump up to: а беременный в дюймовый и Ян Й., Гао Дж., Ван Дж, Хеффернан Р., Хансон Дж., Паливал К., Чжоу Ю (май 2018). «Шестьдесят пять лет долгого марша в прогнозировании вторичной структуры белков: последний отрез?» Полем Брифинги в биоинформатике . 19 (3): 482–494. doi : 10.1093/bib/bbw129 . PMC 5952956 . PMID 28040746 .
- ^ Shasstry Ka, Sanjay Ha (2020). «Машинное обучение для биоинформатики» . В Srinivasa K, Siddesh G, Manisekhar S (Eds.). Статистическое моделирование и принципы машинного обучения для методов биоинформатики, инструментов и приложений . Алгоритмы для интеллектуальных систем. Сингапур: Спрингер. С. 25–39. doi : 10.1007/978-981-15-2445-5_3 . ISBN 978-981-15-2445-5 Полем S2CID 214350490 . Получено 28 июня 2021 года .
- ^ Jump up to: а беременный в Soueidan H, Nikolski M (2019). «Машинное обучение для метагеномики: методы и инструменты». Метагеномика . 1 Arxiv : 1510.06621 . doi : 10.1515/metgen-2016-0001 . ISSN 2449-7657 . S2CID 17418188 .
- ^ Rabiner L, Juang B (январь 1986 г.). «Введение в скрытые модели Маркова» . IEEE ASSP Magazine . 3 (1): 4–16. doi : 10.1109/mass.1986.1165342 . ISSN 1558-1284 . S2CID 11358505 .
- ^ Джексон С.Х., Шарплс Л.Д., Томпсон С.Г., Даффи С.В., Коуто Е (июль 2003 г.). «Multistate Markov Models для прогрессирования заболевания с ошибкой классификации». Журнал Королевского статистического общества, Серия D (Статистик) . 52 (2): 193–209. doi : 10.1111/1467-9884.00351 . S2CID 9824404 .
- ^ Amoros R, King R, Toyoda H, Kumada T, Johnson PJ, Bird TG (30 мая 2019 г.). «Скрытая модель Маркова непрерывного времени с использованием биомаркеров в сыворотке с применением к гепатоцеллюлярной карциноме» . Метрон . 77 (2): 67–86. doi : 10.1007/s40300-019-00151-8 . PMC 6820468 . PMID 31708595 .
- ^ Эдди С.Р. (1 октября 1998 г.). «Профиль скрытые модели Маркова» . Биоинформатика . 14 (9): 755–63. doi : 10.1093/bioinformatics/14.9.755 . PMID 9918945 .
- ^ McClintock BT, Langrock R, Gimenez O, Cam E, Borchers DL, Glennie R, Patterson Ta (декабрь 2020 г.). «Раскрытие динамики экологического состояния со скрытыми моделями Маркова» . Экологические письма . 23 (12): 1878–1903. Arxiv : 2002.10497 . Bibcode : 2020ecoll..23.1878m . doi : 10.1111/ele.13610 . PMC 7702077 . PMID 33073921 .
- ^ Чжан В. (1988). «Нейронная сеть распознавания сдвига и ее оптическая архитектура» . Материалы ежегодной конференции Японского общества прикладной физики .
- ^ Zhang W, Itoh K, Tanida J, Ichioka Y (ноябрь 1990). «Параллельная распределенная модель обработки с локальными инвариантными взаимосвязаниями и ее оптической архитектурой». Прикладная оптика . 29 (32): 4790–7. Bibcode : 1990appt..29.4790z . doi : 10.1364/ao.29.004790 . PMID 20577468 .
- ^ Jump up to: а беременный Епископ, Кристофер М. (17 августа 2006 г.). Распознавание и машинное обучение . Нью -Йорк: Спрингер. ISBN 978-0-387-31073-2 .
- ^ Фукусима К. (2007). «Неокогнитрон» . Scholaredia . 2 (1): 1717. Bibcode : 2007schpj ... 2.1717f . doi : 10.4249/Scholaredia.1717 .
- ^ Hubel DH, Wiesel TN (март 1968 г.). «Рецептивные поля и функциональная архитектура полосатой коры обезьяны» . Журнал физиологии . 195 (1): 215–43. doi : 10.1113/jphysiol.1968.sp008455 . PMC 1557912 . PMID 4966457 .
- ^ Фукусима К. (1980). «Неокогнитрон: модель нейронной сети для самостоятельной организации для механизма распознавания закономерностей, не влияющих на сдвиг в положении». Биологическая кибернетика . 36 (4): 193–202. doi : 10.1007/bf00344251 . PMID 7370364 . S2CID 206775608 .
- ^ Matsugu M, Mori K, Mitari Y, Kaneda Y (2003). «Субъект независимый распознавание выражения лица с надежным обнаружением лица с использованием сверточной нейронной сети». Нейронные сети . 16 (5–6): 555–9. doi : 10.1016/s0893-6080 (03) 00115-1 . PMID 12850007 .
- ^ Fioravanti D, Giarratano Y, Maggio V, Agostinelli C, Chierici M, Jurman G, Furlanelo C (март 2018 г.). «Фиогнотические сверточные нейронные сети в метагеномике» . BMC Bioinformatics . 19 Suppl 2) ( : 49 . PMC 5850953 . PMID 29536822 .
- ^ Джи, Янронг; Чжоу, Чжихан; Лю, Хан; Давулури, Рамана V (9 августа 2021 г.). Келсо, Джанет (ред.). «Днаберт: предварительно обученные двунаправленные представления энкодера из модели трансформаторов для языка ДНК в геноме» . Биоинформатика . 37 (15): 2112–2120. doi : 10.1093/bioinformatics/btab083 . ISSN 1367-4803 . PMC 11025658 . PMID 33538820 .
- ^ Гюндуз, Хюсеин Анил; Биндер, Мартин; К, Сяо-Инь; MRECHES, Рене; Бишл, Бернд; McHardy, Alice C.; Мюнч, Филипп с.; Резаи, Мина (11 сентября 2023 г.). «Самоподобный метод глубокого обучения для эффективного обучения данных по геномике» . Биология связи . 6 (1): 928. doi : 10.1038/s42003-023-05310-2 . ISSN 2399-3642 . PMC 10495322 . PMID 37696966 .
- ^ Ho TK (1995). Случайные решающие леса . Материалы 3 -й Международной конференции по анализу и признанию документов, Монреаль, QC, 14–16 августа 1995 г. с. 278–282.
- ^ Диттерх Т. (2000). Экспериментальное сравнение трех методов для построения ансамблей деревьев решений: сумки, повышение и рандомизация . Kluwer Academic Publishers. С. 139–157.
- ^ Брейман, Лео (2001). "Радовые леса" . Машинное обучение . 45 (1): 5–32. Bibcode : 2001machl..45 .... 5b . doi : 10.1023/a: 1010933404324 . S2CID 89141 .
- ^ Jump up to: а беременный Zhang C, Ma Y (2012). Ансамблевое машинное обучение: методы и приложения . Нью -Йорк: Springer New York Dordrecht Heidelberg London. С. 157–175. ISBN 978-1-4419-9325-0 .
- ^ Карим М.Р., Бейан О., Заппа А., Коста Иг, Ребхольц-Шуманн Д., Кочез М., Декер С. (январь 2021 г.). «Подходы кластеризации на основе глубокого обучения для биоинформатики» . Брифинги в биоинформатике . 22 (1): 393–415. doi : 10.1093/bib/bbz170 . PMC 7820885 . PMID 32008043 .
- ^ Lorbeer B, Kosareva A, Deva B, Softić D, Ruppel P, Küpper A (1 марта 2018 г.). «Изменения на алгоритме кластеризации березы» . Исследование больших данных . 11 : 44–53. doi : 10.1016/j.bdr.2017.09.002 .
- ^ Navarro-Muñoz JC, Selem-Mojica N, Mullowney MW, Kautsar SA, Tryon JH, Parkinson EI, et al. (Январь 2020 г.). «Вычислительная структура для изучения крупномасштабного биосинтетического разнообразия» . Природная химическая биология . 16 (1): 60–68. doi : 10.1038/s41589-019-0400-9 . PMC 6917865 . PMID 31768033 .
- ^ Jump up to: а беременный в дюймовый и Shasstry Ka, Sanjay Ha (2020). «Машинное обучение для биоинформатики». Статистическое моделирование и принципы машинного обучения для методов биоинформатики, инструментов и приложений . Алгоритмы для интеллектуальных систем. Спрингер Сингапур. С. 25–39. doi : 10.1007/978-981-15-2445-5_3 . ISBN 978-981-15-2444-8 Полем S2CID 214350490 .
- ^ Libbrecht MW, Noble WS (июнь 2015 г.). «Приложения машинного обучения в генетике и геномике» . Природные обзоры. Генетика . 16 (6): 321–32. doi : 10.1038/nrg3920 . PMC 5204302 . PMID 25948244 .
- ^ Degroeve S, De Baets B, Van de Peer Y, Rouzé P (2002). «Выбор подмножества функций для прогнозирования сайта сплайсинга» . Биоинформатика . 18 (Suppl 2): S75-83. doi : 10.1093/bioinformatics/18.suppl_2.s75 . PMID 12385987 .
- ^ Huang S, Cai N, Pacheco PP, Warrandes S, Wang Y, Xu W (январь 2018 г.). «Применение поддержки векторной машины (SVM) в геномике рака» . Рак геномика и протеомика . 15 (1): 41–51. doi : 10.21873/cgp.20063 . PMC 5822181 . PMID 29275361 .
- ^ Angermueller C, Pärnamaa T, Parts L, Stegle O (июль 2016 г.). «Глубокое обучение для вычислительной биологии» . Биология молекулярных систем . 12 (7): 878. doi : 10.15252/msb.20156651 . PMC 4965871 . PMID 27474269 .
- ^ Cao C, Liu F, Tan H, Song D, Shu W, Li W, et al. (Февраль 2018 г.). «Глубокое обучение и его применение в биомедицине» . Геномика, протеомика и биоинформатика . 16 (1): 17–32. doi : 10.1016/j.gpb.2017.07.003 . PMC 6000200 . PMID 29522900 .
- ^ Zou J, Huss M, Abid A, Mohammadi P, Torkamani A, Telenti A (январь 2019). «Ученик по глубокому обучению в геномике» . Природа генетика . 51 (1): 12–18. doi : 10.1038/s41588-018-0295-5 . PMC 11180539 . PMID 30478442 . S2CID 205572042 .
- ^ Zeng Z, Shi H, Wu Y, Hong Z (2015). «Обзор методов обработки естественного языка в биоинформатике» . Вычислительные и математические методы в медицине . 2015 (D1): 674296. DOI : 10.1155/2015/674296 . PMC 4615216 . PMID 26525745 .
- ^ Zeng Z, Shi H, Wu Y, Hong Z (2012). «Обзор методов обработки естественного языка в биоинформатике» . Вычислительные и математические методы в медицине . 2015 (D1): 674296. DOI : 10.1016/B978-0-12-385467-4.00006-3 . PMC 4615216 . PMID 26525745 .
- ^ Zeng Z, Shi H, Wu Y, Hong Z (2017). «Обзор методов обработки естественного языка в биоинформатике» . Вычислительные и математические методы в медицине . 2015 (D1): 674296. DOI : 10.1155/2015/674296 . PMC 4615216 . PMID 26525745 .
- ^ «Статистика GenBank и WGS» . www.ncbi.nlm.nih.gov . Получено 25 ноября 2023 года .
- ^ Jump up to: а беременный в дюймовый Мате С., Сагот М.Ф., Шикс Т., Рузе П (октябрь 2002 г.). «Современные методы прогнозирования генов, их сильные и слабые стороны» . Исследование нуклеиновых кислот . 30 (19): 4103–17. doi : 10.1093/nar/gkf543 . PMC 140543 . PMID 12364589 .
- ^ Pratas D, Silva RM, Pinho AJ, Ferreira PJ (май 2015). «Метод без выравнивания для поиска и визуализации перестройки между парами последовательностей ДНК» . Научные отчеты . 5 (10203): 10203. Bibcode : 2015natsr ... 510203p . doi : 10.1038/srep10203 . PMC 4434998 . PMID 25984837 .
- ^ Полинг Л., Кори Р.Б., Брэнсон Хр (апрель 1951 г.). «Структура белков; две связанные с водородными спиральными конфигурациями полипептидной цепи» . Труды Национальной академии наук Соединенных Штатов Америки . 37 (4): 205–11. Bibcode : 1951pnas ... 37..205p . doi : 10.1073/pnas.37.4.205 . PMC 1063337 . PMID 14816373 .
- ^ Jump up to: а беременный Wang S, Peng J, Ma J, Xu J (январь 2016 г.). «Прогнозирование вторичной структуры белка с использованием глубоких сверточных нейронных полей» . Научные отчеты . 6 : 18962. Arxiv : 1512.00843 . BIBCODE : 2016NATSR ... 618962W . doi : 10.1038/srep18962 . PMC 4707437 . PMID 26752681 .
- ^ Riesenfeld CS, Schloss PD, Handelsman J (2004). «Метагеномика: геномный анализ микробных сообществ». Ежегодный обзор генетики . 38 (1): 525–52. doi : 10.1146/annurev.genet.38.072902.091216 . PMID 15568985 .
- ^ Соуидан, Хейссам; Никольски, Мача (1 января 2017 г.). «Машинное обучение для метагеномики: методы и инструменты» . Метагеномика . 1 (1). Arxiv : 1510.06621 . doi : 10.1515/metgen-2016-0001 . ISSN 2449-7657 . S2CID 17418188 .
- ^ Jump up to: а беременный в Лин Й., Ван Г., Ю., Сун Дж.Дж. (апрель 2021 г.). «Искусственный интеллект и метагеномика при кишечных заболеваниях» . Журнал гастроэнтерологии и гепатологии . 36 (4): 841–847. doi : 10.1111/jgh.15501 . PMID 33880764 . S2CID 233312307 .
- ^ Jump up to: а беременный в Данг Т, Кишино Х (январь 2020 г.). «Обнаружение значительных компонентов микробиомов случайным лесом с прямой переменной отбором и филогенетикой». Biorxiv 10.1101/2020.10.29.361360 .
- ^ Fioravanti D, Giarratano Y, Maggio V, Agostinelli C, Chierici M, Jurman G, Furlanelo C (март 2018 г.). «Фиогнотические сверточные нейронные сети в метагеномике» . BMC Bioinformatics . 19 Suppl 2) ( : 49 . PMC 5850953 . PMID 29536822 .
- ^ Dhungel E, Mreyoud Y, Gwak HJ, Rajeh A, Rho M, Ahn Th (январь 2021 г.). «Мегар: интерактивный пакет R для быстрой классификации образцов и прогнозирования фенотипа с использованием профилей метагенома и машинного обучения» . BMC Bioinformatics . 22 (1): 25. doi : 10.1186/s12859-020-03933-4 . PMC 7814621 . PMID 33461494 .
- ^ Xun W, Liu Y, Li W, Ren Y, Xiong W, Xu Z, et al. (Январь 2021 г.). «Специализированные метаболические функции таксонов Keystone поддерживают стабильность микробиома почвы» . Микробиом . 9 (1): 35. doi : 10.1186/s40168-020-00985-9 . PMC 7849160 . PMID 33517892 .
- ^ Jump up to: а беременный Pirooznia M, Yang JY, Yang MQ, Deng Y (2008). «Сравнительное исследование различных методов машинного обучения по данным экспрессии генов микрочипа» . BMC Genomics . 9 Suppl 1 (1): S13. doi : 10.1186/1471-2164-9-S1-S13 . PMC 2386055 . PMID 18366602 .
- ^ «Машинное обучение в биологии молекулярных систем» . Границы . Получено 9 июня 2017 года .
- ^ D'Alché-Buc F, Wehenkel L (декабрь 2008 г.). «Машинное обучение в системной биологии» . BMC COURTINGINGS . 2 Suppl 4 (4): S1. doi : 10.1186/1753-6561-2-S4-S1 . PMC 2654969 . PMID 19091048 .
- ^ Бхаттачарья М (2020). «Неконтролируемые методы в геномике». В Srinivasa MG, Siddesh GM, Manisekhar Sr (ред.). Статистическое моделирование и принципы машинного обучения для методов биоинформатики, инструментов и приложений . Спрингер Сингапур. С. 164–188. ISBN 978-981-15-2445-5 .
- ^ Topol EJ (январь 2019 г.). «Высокопроизводительная медицина: конвергенция человеческого и искусственного интеллекта». Природная медицина . 25 (1): 44–56. doi : 10.1038/s41591-018-0300-7 . PMID 30617339 . S2CID 57574615 .
- ^ Jump up to: а беременный Цзян Ф., Цзян Ю., Чжи Х, Донг Й, Ли Х, Мас и др. (Декабрь 2017). «Искусственный интеллект в здравоохранении: прошлое, настоящее и будущее» . Инсульт и сосудистая неврология . 2 (4): 230–243. doi : 10.1136/svn-2017-000101 . PMC 5829945 . PMID 29507784 .
- ^ Mirtskhulava L, Wong J, Al-Majeed S, Pearce G (март 2015 г.). «Модель искусственной нейронной сети в диагностике инсульта» (PDF) . 17-я Международная конференция UKSIM-AMSS по моделированию и моделированию (UKSIM) . С. 50–53. doi : 10.1109/uksim.2015.33 . ISBN 978-1-4799-8713-9 Полем S2CID 6391733 .
- ^ Титано Дж.Дж., Баджли М., Шеффлейн Дж., Пейн М., Су А., Кай М. и др. (Сентябрь 2018 г.). «Автоматизированное наблюдение за глубокими сетевыми сетью для черепных изображений для острых неврологических событий». Природная медицина . 24 (9): 1337–1341. doi : 10.1038/s41591-018-0147-y . PMID 30104767 . S2CID 51976344 .
- ^ Jump up to: а беременный в Краллингер М., Эрхардт Р.А., Валенсия А (март 2005 г.). «Текстовые подходы в молекулярной биологии и биомедицине». Drug Discovery сегодня . 10 (6): 439–45. doi : 10.1016/s1359-6446 (05) 03376-3 . PMID 15808823 .
- ^ Pratas D, Hosseini M, Silva R, Pinho A, Ferreira P (20–23 июня 2017 г.). «Визуализация различных областей ДНК современного человека относительно неандертальского генома». Распознавание шаблона и анализ изображений . Заметки лекции в информатике. Тол. 10255. С. 235–242. doi : 10.1007/978-3-319-58838-4_26 . ISBN 978-3-319-58837-7 .
- ^ Bardgett Rd, Caruso T (март 2020 г.). «Ответы микробного сообщества почвы на крайности климата: сопротивление, устойчивость и переходы к альтернативным состояниям» . Философские транзакции Королевского общества Лондона. Серия B, биологические науки . 375 (1794): 20190112. DOI : 10.1098/rstb.2019.0112 . PMC 7017770 . PMID 31983338 .
- ^ Deveau A, Bonito G, Uehling J, Paoletti M, Becker M, Bindschedler S, et al. (Май 2018). «Бактериальногенские взаимодействия: экология, механизмы и проблемы» . Обзоры микробиологии FEMS . 42 (3): 335–352. doi : 10.1093/femsre/fuy008 . HDL : 21.11116/0000-0002-C1E7-F . PMID 29471481 .
- ^ Ансари М.З, Ядав Г., Гохале Р.С., Моханти Д. (июль 2004 г.). «NRPS-PKS: ресурс, основанный на знаниях для анализа NRPS/PKS Megasynthases» . Исследование нуклеиновых кислот . 32 (Проблема веб-сервера): W405-13. doi : 10.1093/nar/gkh359 . PMC 441497 . PMID 15215420 .
- ^ Jump up to: а беременный Navarro-Muñoz JC, Selem-Mojica N, Mullowney MW, Kautsar SA, Tryon JH, Parkinson EI, et al. (Январь 2020 г.). «Вычислительная структура для изучения крупномасштабного биосинтетического разнообразия» . Природная химическая биология . 16 (1): 60–68. doi : 10.1038/s41589-019-0400-9 . PMC 6917865 . PMID 31768033 .
- ^ Jump up to: а беременный Doroghazi JR, Albright JC, Geering AW, Ju KS, Haines RR, Tchalukov KA, et al. (Ноябрь 2014). «Дорожная карта для открытия натурального продукта на основе крупномасштабной геномики и метаболомики» . Природная химическая биология . 10 (11): 963–8. doi : 10.1038/nchembio.1659 . PMC 4201863 . PMID 25262415 .
- ^ Jump up to: а беременный Cimermancic P, Medema MH, Claesen J, Kurita K, Wieland Brown Lc, Mavrommatis K, et al. (Июль 2014). «Понимание вторичного метаболизма из глобального анализа прокариотических биосинтетических генов» . Клетка . 158 (2): 412–421. doi : 10.1016/j.cell.2014.06.034 . PMC 4123684 . PMID 25036635 .
- ^ Jump up to: а беременный Geering AW, McClure RA, Doroghazi JR, Albright JC, Haverland NA, Zhang Y, et al. (Февраль 2016 г.). «Метабологеномика: корреляция кластеров микробных генов с метаболитами приводит к открытию нерибосомного пептида с необычным аминокислотным мономером» . ACS Central Science . 2 (2): 99–108. doi : 10.1021/acscentsci.5b00331 . PMC 4827660 . PMID 27163034 .
- ^ Amiri Moghaddam J, Crüsemann M, Alanjary M, Harms H, Dávila-Céspedes A, Blom J, et al. (Ноябрь 2018). «Анализ генома и метаболом морских миксобактерий выявляет высокий потенциал для биосинтеза новых специализированных метаболитов» . Научные отчеты . 8 (1): 16600. BIBCODE : 2018NATSR ... 8166600A . doi : 10.1038/s41598-018-34954-y . PMC 6226438 . PMID 30413766 .
- ^ Дункан К.Р., Крюсеман М., Лехнер А., Саркар А., Ли Дж, Зимерт Н. и др. (Апрель 2015). «Молекулярная сеть и добыча генома на основе рисунков улучшает открытие биосинтетических генов и их продуктов от видов Salinispora» . Химия и биология . 22 (4): 460–471. doi : 10.1016/j.chembiol.2015.03.010 . PMC 4409930 . PMID 25865308 .
- ^ Nielsen JC, Grijseels S, Pririgent S, Ji B, Dainat J, Nielsen KF, et al. (Апрель 2017). «Глобальный анализ биосинтетических кластеров генов выявляет огромный потенциал вторичного выработки метаболита у видов Penicillium». Природная микробиология . 2 (6): 17044. DOI : 10.1038/nmicrobiol.2017.44 . PMID 28368369 . S2CID 22699928 .
- ^ McClure RA, Geering AW, JU KS, Baccile JA, Schroeder FC, Metcalf WW, et al. (Декабрь 2016 г.). «Выяснение семейств природных продуктов римозамида-детоксина и их биосинтез с использованием корреляций метаболита/генов» . ACS Химическая биология . 11 (12): 3452–3460. doi : 10.1021/acschembio.6b00779 . PMC 5295535 . PMID 27809474 .
- ^ Cao L, Shcherbin E, Mohimani H (август 2019). «Сеть ассоциации в общении метаболом и метагенома выявляет микробные натуральные продукты и микробные биотрансформационные продукты от микробиоты человека» . Msystems . 4 (4): E00387–19, /msystems/4/4/msys.00387–19.atom. doi : 10.1128/msystems.00387-19 . PMC 6712304 . PMID 31455639 .
- ^ Olm MR, Bhattacharya N, Crits-Christoph A, Firek BA, Baker R, Song YS, et al. (Декабрь 2019). «Некротизирующему энтероколиту предшествует повышенная репликация бактерий кишечника, Klebsiella и бактерии, кодирующие Fimbriae» . Наука достижения . 5 (12): eaax5727. Bibcode : 2019scia .... 5.5727o . doi : 10.1126/sciadv.aax5727 . PMC 6905865 . PMID 31844663 .
- ^ Carrión VJ, Perez-Jaramillo J, Cordovez V, Tracanna V, De Hollander M, Ruiz-Buck D, et al. (Ноябрь 2019). «Индуцированная патогеном активация функций, вызванных заболеванием, в микробиоме эндофитного корня» . Наука . 366 (6465): 606–612. Bibcode : 2019sci ... 366..606c . doi : 10.1126/science.aaw9285 . HDL : 1887/3188901 . PMID 31672892 . S2CID 207814746 .
- ^ Паскаль Андреу, Виктория; Augustijn, Hannah E.; Ван ден Берг, Коэн; Ван дер Хофт, Джастин Дж.Дж; Фишбах, Майкл А.; Medema, Marnix H. (26 октября 2021 г.). Шанк, Элизабет Энн (ред.). «Большая папка: автоматизированный трубопровод для профиля метаболического кластера численности и экспрессии в микробиомах» . Msystems . 6 (5): E0093721. Doi : 10.1128/msystems.00937-21 . ISSN 2379-5077 . PMC 8547482 . PMID 34581602 .
- ^ Agrawal P, Khater S, Gupta M, Sain N, Mohanty D (июль 2017 г.). «Rippminer: ресурс биоинформатики для расшифровки химических структур RIPPS на основе прогнозирования расщепления и поперечных связей» . Исследование нуклеиновых кислот . 45 (W1): W80 - W88. doi : 10.1093/nar/gkx408 . PMC 5570163 . PMID 28499008 .
- ^ Huber F, Ridder L, Verhoeven S, Spaaks JH, Diblen F, Rogers S, Van Der Hooft JJ (февраль 2021 г.). «Spec2VEC: улучшение сходства масс -спектра за счет изучения структурных отношений» . PLOS Computational Biology . 17 (2): E1008724. BIBCODE : 2021PLSCB..17E8724H . doi : 10.1371/journal.pcbi.1008724 . PMC 7909622 . PMID 33591968 .
- ^ Huber F, Ridder L, Verhoeven S, Spaaks JH, Diblen F, Rogers S, Van Der Hooft JJ (февраль 2021 г.). «Spec2VEC: улучшение сходства масс -спектра за счет изучения структурных отношений» . PLOS Computational Biology . 17 (2): E1008724. BIBCODE : 2021PLSCB..17E8724H . doi : 10.1371/journal.pcbi.1008724 . PMC 7909622 . PMID 33591968 .
- ^ Национальный центр информации о биотехнологии; Национальная библиотека медицины США. «Национальный центр биотехнологической информации» . ncbi.nlm.nih.gov . Получено 30 июля 2021 года .
- ^ Agarwala R, Barrett T, Beck J, Benson DA, Bollin C, Bolton E, et al. (Координаторы ресурсов NCBI) (январь 2018 г.). «Ресурсы базы данных Национального центра биотехнологической информации» . Исследование нуклеиновых кислот . 46 (D1): D8 - D13. doi : 10.1093/nar/gkx1095 . PMC 5753372 . PMID 29140470 .
- ^ «База данных Antismash» . Antismash-DB.Se-SecondaryMetabolites.org .
- ^ «Мибиг: минимальная информация о биосинтетическом кластере генов» . mibig.se -secondarymetabolites.org . Получено 30 июля 2021 года .
- ^ Любовь
- ^ Пример в, Blin K, Shaw S, Navarro-Muñoz JC. (Январь 2020 г.). "Мибибиг 2.0 : нуклеиновых кислот Исследование 48 (D1): D454 - D458. doi : 10.1093/ nar/ gkz8 PMC 7145714 . PMID 31612915 .
- ^ "Сильва" . Arb-silva.de . Получено 30 июля 2021 года .
- ^ Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. (Январь 2013). «Проект базы данных рибосомальных генов рибосом Silva: улучшенная обработка данных и веб-инструменты» . Исследование нуклеиновых кислот . 41 (выпуск базы данных): D590-6. doi : 10.1093/nar/gks1219 . PMC 3531112 . PMID 23193283 .
- ^ "greengenes.secondgenome.com" . greenges.secondgenome.com . Получено 30 июля 2021 года .
- ^ Desantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. (Июль 2006 г.). «Greengenes, база данных гена 16S RRNA 16S 16S и рабочая панель, совместимая с ARB» . Прикладная и экологическая микробиология . 72 (7): 5069–72. Bibcode : 2006apenm..72.5069d . doi : 10.1128/aem.03006-05 . PMC 1489311 . PMID 16820507 .
- ^ McDonald D, Price MN, Goodrich J, Nawrocki EP, Desantis TZ, Probst A, et al. (Март 2012 г.). «Улучшенная таксономия Грингена с явными рядами для экологического и эволюционного анализа бактерий и археи» . Журнал ISME . 6 (3): 610–8. Bibcode : 2012ismej ... 6..610M . doi : 10.1038/ismej.2011.139 . PMC 3280142 . PMID 22134646 .
- ^ "Opentree" . Tree.opentreeoflife.org . Получено 30 июля 2021 года .
- ^ Hinchliff CE, Smith SA, Allman JF, Burleigh JG, Chaudhary R, Coghill LM, et al. (Октябрь 2015). «Синтез филогения и таксономии в всеобъемлющее дерево жизни» . Труды Национальной академии наук Соединенных Штатов Америки . 112 (41): 12764–9. Bibcode : 2015pnas..11212764H . doi : 10.1073/pnas.1423041112 . PMC 4611642 . PMID 26385966 .
- ^ «RDP -выпуск 11 - инструменты анализа последовательности» . rdp.cme.msu.edu . Архивировано из оригинала 19 августа 2020 года . Получено 30 июля 2021 года .
- ^ Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. (Январь 2014). «Проект рибосомной базы данных: данные и инструменты для анализа высокой пропускной способности РРНК» . Исследование нуклеиновых кислот . 42 (проблема базы данных): D633-42. doi : 10.1093/nar/gkt1244 . PMC 3965039 . PMID 24288368 .
Дифференцируемые вычисления |
|---|