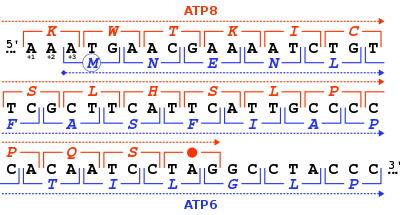
Расположение гена MT-ATP8 в митохондриальном геноме человека. MT-ATP8 является одним из двух митохондриальных генов АТФ-синтазы (красные прямоугольники). 46-нуклеотидное перекрытие в рамках считывания митохондриальных генов человека MT-ATP8 и MT-ATP6 . Для каждого триплета нуклеотидов (квадратные скобки) указана соответствующая аминокислота (однобуквенный код) либо в рамке +1 для MT-ATP8 (красный цвет), либо в рамке +3 для MT-ATP6 (синий цвет). .
MT-ATP8 (или ATP8 ) представляет собой митохондриальный ген с полным названием «митохондриально кодируемая субъединица 8 мембраны АТФ-синтазы», который кодирует субъединицу митохондриальной АТФ-синтазы , -синтазы АТФ субъединицу 8 (или субъединицу A6L ). Эта субъединица принадлежит к комплексу F o большой трансмембранной АТФ-синтазы F-типа . [5] Этот фермент, также известный как комплекс V, отвечает за заключительный этап окислительного фосфорилирования в цепи переноса электронов . В частности, один сегмент АТФ-синтазы позволяет положительно заряженным ионам , называемым протонами , проходить через специализированную мембрану внутри митохондрий. Другой сегмент фермента использует энергию, создаваемую этим потоком протонов, для преобразования молекулы, называемой аденозиндифосфатом (АДФ), в АТФ . [6] Субъединица 8 различается по последовательности у Metazoa , растений и грибов .
АТФ-синтазы человека и других млекопитающих кодируется в митохондриальном геноме геном MT-ATP8 Белок 8 . Когда полный митохондриальный геном человека был впервые опубликован, ген MT-ATP8 был описан как неидентифицированная рамка считывания URF A6L . [5] Необычной особенностью гена MT-ATP8 является его 46-нуклеотидное перекрытие с геном MT-ATP6 . Что касается рамки считывания (+1) MT-ATP8 , ген MT-ATP6 начинается в рамке считывания +3.
Белок MT-ATP8 весит 8 кДа и состоит из 68 аминокислот . [7] [8] Белок является субъединицей F 1 F o АТФазы, также известной как Комплекс V , который состоит из 14 ядерно- и 2 митохондриально-кодируемых субъединиц. АТФазы F-типа состоят из двух структурных доменов: F 1 , содержащего внемембранное каталитическое ядро, и F o , содержащего мембранный протонный канал, соединенных между собой центральным стеблем и периферическим стеблем. В качестве субъединицы А MT-ATP8 содержится в некаталитической трансмембранной части F o комплекса, включающей протонный канал . Каталитическая часть митохондриальной АТФ-синтазы состоит из 5 различных субъединиц (альфа, бета, гамма, дельта и эпсилон), собранных со стехиометрией 3 альфа, 3 бета и одного представителя остальных 3. Протонный канал состоит из трех основные субъединицы (а, б, в). Этот ген кодирует дельта-субъединицу каталитического ядра. Были идентифицированы альтернативно сплайсированные варианты транскриптов, кодирующие одну и ту же изоформу. [9] [6]
Эта белковая субъединица, по-видимому, является неотъемлемым компонентом статорной ножки дрожжей митохондриальных F-АТФаз . [11] Стебель статора закреплен в мембране и предотвращает бесполезное вращение субъединиц АТФазы относительно ротора во время сопряженного синтеза/гидролиза АТФ. Эта субъединица может иметь аналогичную функцию у Metazoa .
Номенклатура . фермента имеет давнюю историю Фракция F 1 получила свое название от термина «Фракция 1», а F o (записанная как буква «о», а не «ноль») получила свое название от того, что она является связывающей фракцией для олигомицина , типа антибиотика природного происхождения. который способен ингибировать единицу F o АТФ-синтазы. [12] [13] Область F o АТФ-синтазы представляет собой протонную пору, встроенную в мембрану митохондрий. Он состоит из трех основных субъединиц A, B и C и (у человека) шести дополнительных субъединиц: d , e , f , g , MT-ATP6 (или F6) и MT-ATP8 (или A6L). 3D-структуру гомолога этой субъединицы E. coli смоделировали На основе данных электронной микроскопии (цепь М PDB : 1c17 ). Он образует трансмембранный 4-α-пучок.
Мутации MT-ATP8 и других генов, влияющих на окислительное фосфорилирование в митохондриях, связаны с различными нейродегенеративными и сердечно-сосудистыми заболеваниями, включая дефицит митохондриального комплекса V, наследственную оптическую нейропатию Лебера (LHON), митохондриальную энцефаломиопатию с инсультоподобными эпизодами ( MELAS ). и синдром Лея синдром НАРП . Большинство клеток организма содержат тысячи митохондрий, каждая из которых содержит одну или несколько копий митохондриальной ДНК . Тяжесть некоторых митохондриальных нарушений связана с процентом митохондрий в каждой клетке, имеющих определенное генетическое изменение. Люди с синдромом Ли из-за мутации гена MT-ATP6, как правило, имеют очень высокий процент митохондрий с мутацией (от более 90 до 95 процентов). Менее тяжелые проявления NARP обусловлены меньшим процентом митохондрий с мутацией, обычно от 70 до 90 процентов. Поскольку эти два состояния являются результатом одних и тех же генетических изменений и могут возникать у разных членов одной семьи, исследователи полагают, что они могут представлять собой спектр перекрывающихся признаков, а не два отдельных синдрома. [6]
Дефицит митохондриального комплекса V проявляется гетерогенными клиническими проявлениями, включая нейропатию , атаксию , гипертрофическую кардиомиопатию . Гипертрофическая кардиомиопатия может проявляться гипертрофией от незначительной до крайней степени , фиброзом от минимального до обширного и нарушением миоцитов , отсутствием или тяжелой обструкцией выносящего тракта левого желудочка и отчетливыми контурами/морфологией перегородки с чрезвычайно различным клиническим течением. [14] [15]
Дефицит митохондриального комплекса V — это недостаток (дефицит) или потеря функции комплекса V цепи переноса электронов , который может вызывать широкий спектр признаков и симптомов, затрагивающих многие органы и системы организма, особенно нервную систему и сердце . Расстройство может быть опасным для жизни в младенчестве или раннем детстве. У больных могут возникнуть проблемы с питанием, медленный рост, низкий мышечный тонус ( гипотония ), сильная утомляемость ( вялость ) и задержка развития . У них наблюдается тенденция к повышению уровня молочной кислоты в крови ( лактоацидоз ), что может вызывать тошноту, рвоту, слабость и учащенное дыхание. Высокий уровень аммиака в крови ( гипераммониемия ) также может наблюдаться у больных людей и в некоторых случаях приводит к нарушению функции мозга ( энцефалопатии ) и повреждению других органов. [16] Атаксия , микроцефалия , задержка развития и умственная отсталость наблюдались у пациентов с мутацией сдвига рамки считывания в MT-ATP6. Это вызывает вставку C в положении 8612, что приводит к укороченному белку длиной всего 36 аминокислот, и два однонуклеотидных полиморфизма T > C в положениях 8610 и 8614, которые приводят к гомополимерному растяжению цитозина . [17]
Гипертрофическая кардиомиопатия , распространенная особенность дефицита митохондриального комплекса V, характеризуется утолщением ( гипертрофией ) сердечной мышцы , что может привести к сердечной недостаточности . [16] Мутация m.8528T>C возникает в перекрывающейся области генов MT-ATP6 и MT-ATP8 и была описана у многих пациентов с детской кардиомиопатией. Эта мутация меняет инициирующий кодон в MT-ATP6 на треонин, а также заменяет триптофан на аргинин в положении 55 MT-ATP8. [18] [15] Лица с дефицитом митохондриального комплекса V также могут иметь характерные черты лица, включая высокий лоб, изогнутые брови, направленные вниз внешние уголки глаз (наклонные глазные щели ), выступающую переносицу, низко посаженные уши, тонкие губы и маленький подбородок ( микрогнатия ). [16]
^ Jump up to: а б с Уэр С.М., Эль-Хасан Н., Калер С.Г., Чжан К., Ма Ю.В., Миллер Э., Вонг Б., Спайсер Р.Л., Крейген В.Дж., Козел Б.А., Грейндж Д.К., Вонг Л.Дж. (май 2009 г.). «Детская кардиомиопатия, вызванная мутацией в перекрывающейся области генов митохондриальной АТФазы 6 и 8». Журнал медицинской генетики . 46 (5): 308–14. дои : 10.1136/jmg.2008.063149 . ПМИД 19188198 . S2CID 25354118 .
Торрони А., Ахилли А., Маколей В., Ричардс М., Бандельт Х.Дж. (июнь 2006 г.). «Сбор плодов дерева мтДНК человека». Тенденции в генетике . 22 (6): 339–45. дои : 10.1016/j.tig.2006.04.001 . PMID 16678300 .
Бодентейх А., Митчелл Л.Г., Полимеропулос М.Х., Меррил Ч.Р. (май 1992 г.). «Динуклеотидный повтор в D-петле митохондрий человека». Молекулярная генетика человека . 1 (2): 140. doi : 10.1093/hmg/1.2.140-a . ПМИД 1301157 .
Лу X, Уокер Т., Макманус Дж.П., Селиги В.Л. (июль 1992 г.). «Дифференциация клеток аденокарциномы толстой кишки человека HT-29 коррелирует с повышенной экспрессией митохондриальной РНК: влияние трегалозы на рост и созревание клеток». Исследования рака . 52 (13): 3718–25. ПМИД 1377597 .
Марзуки С., Ноер А.С., Лертрит П., Тьягараджан Д., Капса Р., Уттанафол П., Бирн Э. (декабрь 1991 г.). «Нормальные варианты митохондриальной ДНК человека и продукты трансляции: построение справочной базы данных». Генетика человека . 88 (2): 139–45. дои : 10.1007/bf00206061 . ПМИД 1757091 . S2CID 28048453 .
Arc.Ask3.Ru Номер скриншота №: ea9501b6a57656c6c8c4e0f6477dc0bd__1713154200 URL1:https://arc.ask3.ru/arc/aa/ea/bd/ea9501b6a57656c6c8c4e0f6477dc0bd.html Заголовок, (Title) документа по адресу, URL1: MT-ATP8 - Wikipedia
Данный printscreen веб страницы (снимок веб страницы, скриншот веб страницы), визуально-программная копия документа расположенного по адресу URL1 и сохраненная в файл, имеет: квалифицированную, усовершенствованную (подтверждены: метки времени, валидность сертификата), открепленную ЭЦП (приложена к данному файлу), что может быть использовано для подтверждения содержания и факта существования документа в этот момент времени. Права на данный скриншот принадлежат администрации Ask3.ru, использование в качестве доказательства только с письменного разрешения правообладателя скриншота. Администрация Ask3.ru не несет ответственности за информацию размещенную на данном скриншоте. Права на прочие зарегистрированные элементы любого права, изображенные на снимках принадлежат их владельцам. Качество перевода предоставляется как есть. Любые претензии, иски не могут быть предъявлены. Если вы не согласны с любым пунктом перечисленным выше, вы не можете использовать данный сайт и информация размещенную на нем (сайте/странице), немедленно покиньте данный сайт. В случае нарушения любого пункта перечисленного выше, штраф 55! (Пятьдесят пять факториал, Денежную единицу (имеющую самостоятельную стоимость) можете выбрать самостоятельно, выплаичвается товарами в течение 7 дней с момента нарушения.)



 В эту статью включен текст, доступный по лицензии CC BY 4.0 .
В эту статью включен текст, доступный по лицензии CC BY 4.0 .  В данную статью включен текст из этого источника, находящегося в свободном доступе .
В данную статью включен текст из этого источника, находящегося в свободном доступе .