Малоновая ацидурия
Эта статья нуждается в дополнительных цитатах для проверки . ( июнь 2020 г. ) |
| Малоновая ацидурия | |
|---|---|
| Другие имена | Дефицит малонил-КоА-декарбоксилазы , классическая CMAMMA |
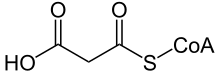 | |
| Малонил-КоА | |
Малоновая ацидурия или недостаточность малонил-КоА декарбоксилазы ( МКД ) является аутосомно - рецессивным заболеванием. [1] нарушение обмена веществ, вызванное генетической мутацией, нарушающей активность малонил-КоА-декарбоксилазы . Этот фермент расщепляет малонил-КоА (предшественник жирных кислот и блокатор окисления жирных кислот) на ацетил-КоА и диоксид углерода .
Признаки и симптомы
[ редактировать ]Признаки и симптомы этого расстройства обычно появляются в раннем детстве. Почти у всех больных детей наблюдается задержка развития. Дополнительные признаки и симптомы могут включать слабый мышечный тонус ( гипотонию ), судороги , диарею , рвоту и низкий уровень сахара в крови ( гипогликемию ). Заболевание сердца, называемое кардиомиопатией , которое ослабляет и увеличивает сердечную мышцу, является еще одной распространенной особенностью малоновой ацидурии.
Генетика
[ редактировать ]
Малоновая ацидурия вызвана мутациями гена MLYCD , расположенного на хромосоме 16q23.3 . [2] Ген кодирует фермент малонил-КоА-декарбоксилазу. Внутри клеток этот фермент помогает регулировать образование и расщепление определенной группы жиров, называемых жирными кислотами .
Многие ткани, включая сердечную мышцу, используют жирные кислоты в качестве основного источника энергии. Мутации в гене MLYCD снижают или устраняют функцию малонил-КоА-декарбоксилазы. Недостаток этого фермента нарушает нормальный баланс образования и распада жирных кислот. В результате жирные кислоты не могут быть преобразованы в энергию, что может привести к характерным особенностям этого заболевания, таким как низкий уровень сахара в крови и кардиомиопатия. Побочные продукты переработки жирных кислот накапливаются в тканях, что также способствует появлению признаков и симптомов малоновой ацидурии.
Малоновая ацидурия наследуется по аутосомно-рецессивному типу. [1] Это означает, что дефектный ген расположен на аутосоме ( хромосома 16 является аутосомой), и чтобы родиться с этим заболеванием, необходимо две копии дефектного гена – по одной унаследованной от каждого родителя. Оба родителя ребенка с аутосомно-рецессивным заболеванием несут по одной копии дефектного гена, но обычно не страдают этим заболеванием.
Малоновая ацидурия встречается крайне редко; данные свидетельствуют о том, что она вызвана нарушением регуляции транскрипции белка. [3] Глядя на молекулярную основу, обнаружено, что две различные гомозиготные мутации вызывают малоновую ацидурию у человека. Первая мутация представляет собой трансверсию гена из C в G, вызывающую преждевременный сигнал остановки в белке. Вторая мутация представляет собой вставку пары оснований в зрелую РНК, что в конечном итоге приводит к усечению белка. [4]
Исследование также подтвердило, что гомозиготная мутация, которая в конечном итоге приводит к малоновой ацидурии, вызвана изодисомией материнской UPD. Это указывает на то, что такое заболевание, вероятно, будет унаследовано от профиля генов матери, а не от отцовского источника. [5]
Распространенность
[ редактировать ]По данным Orphanet (2006), распространенность составляет менее 1 на 1 000 000. [6]
В 1984 году малоновая ацидурия была впервые описана в научном исследовании. [7]
К 1999 г. в Австралии было зарегистрировано только семь случаев малоновой ацидурии у людей; однако этот дефицит преимущественно возникает в детстве. Пациенты из семи зарегистрированных случаев малоновой ацидурии имеют возраст от 4 дней до 13 лет, и все они имеют общий симптом задержки неврологического развития. [4]
К 2006 г. в мировой литературе было опубликовано 17 случаев малоновой ацидурии в возрасте от 8 дней до 12 лет. [3]
К 2017 году в литературе было известно менее 30 случаев. [8]
Патофизиология
[ редактировать ]Декарбоксилаза малонил-КоА действует как катализатор превращения малонил-КоА в ацетил-КоА и CO 2 . [9] Без ферментативной активности малонил-КоА-декарбоксилазы клеточный малонил-КоА увеличивается настолько резко, что в конце вместо этого он расщепляется неспецифической короткоцепочечной ацил-КоА-гидролазой, которая производит малоновую кислоту и КоА. Малоновая кислота является ингибитором цикла Кребса клетками , препятствуя выработке АТФ путем окисления. В этом состоянии клетки, чтобы вырабатывать АТФ, вынуждены усиливать гликолиз, в результате которого в качестве побочного продукта образуется молочная кислота. Увеличение количества молочной и малоновой кислот резко снижает pH крови и вызывает как молочную, так и малоновую ацидурию (кислую мочу). Предполагается также, что избыток митохондриального малонил-КоА повышает уровень метилмалоновой кислоты , что обусловлено ингибирующим действием на метилмалонил-КоА-мутазу . [10] [11]
В цитоплазме малонил-КоА действует как ингибитор фермента карнитинпальмитоилтрансферазы I (CPT1) наружной мембраны митохондрий, который, следовательно, ингибирует окисление жирных кислот . [3] Ингибирующее действие цитопластического малонил-КоА на CPT1 варьируется, поэтому в сердечных и скелетных мышцах он ингибирует примерно в 100 раз сильнее, чем в печени. [12]
Некоторые общие симптомы малоновой ацидурии, такие как кардиомиопатия и метаболический ацидоз, вызваны высокими концентрациями малонил-КоА в цитоплазме.
Хотя мы еще не получили четкого понимания патогенетического механизма этого дефицита, некоторые исследователи предположили, что между малонил-КоА и ферментом CTP1 существует специфическое для мозга взаимодействие, которое может приводить к необъяснимым симптомам малоновой ацидурии. [5]
Исследования показали, что большое количество малонил-КоА-карбоксилазы открепляется в гипоталамусе и коре головного мозга, где обнаруживаются высокие уровни липогенных ферментов, что указывает на то, что малонил-КоА-декарбоксилаза играет роль в синтезе липидов в мозге. [3] Нарушение взаимодействия между малонил-КоА и CPT1 также может способствовать аномальному развитию мозга. [3]
Малонил-КоА-декарбоксилаза играет важную роль в процессах β-окисления как в митохондриях, так и в пероксисомах. [4] Некоторые другие авторы также предположили, что именно дефицит малонил-КоА-карбоксилазы, индуцированный ингибированием пероксисомального β-окисления, способствует задержке развития. [4]
Диагностика
[ редактировать ]Скрининг на повышенный уровень органических кислот, особенно малоновой кислоты и метилмалоновой кислоты, а также на высокий уровень малонилкарнитина . [6] Диагноз затем может быть подтвержден путем определения активности фермента малонил-КоА-декарбоксилазы в культивированных фибробластах кожи. [6] Молекулярно-генетическое тестирование гена MLYCD также может быть полезным. [6]
Дифференциальный диагноз
[ редактировать ]Комбинированная малоновая и метилмалоновая ацидурия (CMAMMA) и классическая метилмалоновая ацидемия
[ редактировать ]Подсчитав соотношение малоновой и метилмалоновой кислот в плазме крови, малоновую ацидурию можно четко отличить от комбинированной малоновой и метилмалоновой ацидурии (CMAMMA) и классической метилмалоновой ацидемии . [13] Последнее применимо как к людям, реагирующим на витамин B 12 , так и к не отвечающим на него при метилмалоновой ацидемии. [13] При малоновой ацидурии результаты соотношения больше 1, так как значение малоновой кислоты выше, чем значение метилмалоновой кислоты. [3] С другой стороны, в CMAMMA результат меньше 1, поскольку содержание метилмалоновой кислоты выше, чем малоновой кислоты. [10]
Уход
[ редактировать ]Исследование, проведенное в Нидерландах, показало, что добавки с карнитином и диета с низким содержанием жиров могут помочь снизить уровень малоновой кислоты в нашем организме. [5]
Примечание
[ редактировать ]Поскольку при малоновой ацидурии повышены уровни как малоновой, так и метилмалоновой кислоты, раньше ее называли комбинированной малоновой и метилмалоновой ацидурией (CMAMMA). Хотя дефицит ACSF3 был обнаружен позднее, термин «комбинированная малоновая и метилмалоновая ацидурия» теперь стал общепринятым в медицинских базах данных для обозначения дефицита ACSF3. [14] [15]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Jump up to: а б Макфи, Великобритания; Логан, RW; Митчелл, Дж. С.; Хауэллс, Д.В.; Цоцис, Э.; Торберн, ДР (октябрь 1993 г.). «Малонил-коэнзим и дефицит декарбоксилазы» . Архив болезней в детстве . 69 (4): 433–436. дои : 10.1136/adc.69.4.433 . ПМК 1029550 . ПМИД 8259873 .
- ^ Интернет-менделевское наследование у человека (OMIM): 606761
- ^ Jump up to: а б с д и ж де Вит МК, де Ку ИФ, Вербек Э, Шот Р., Шондервурд ГК, Дюран М, де Клерк Дж.Б., Хейманс Дж.Г., Лекин М.Х., Верхейен Ф.В., Манчини GM (февраль 2006 г.). «Нарушения головного мозга при дефиците малонил-КоА-декарбоксилазы». Молекулярная генетика и обмен веществ . 87 (2): 102–6. дои : 10.1016/j.ymgme.2005.09.009 . ПМИД 16275149 .
- ^ Jump up to: а б с д ФитцПатрик, доктор медицинских наук; Хилл, А; Толми, Дж.Л.; Торберн, ДР; Христодулу, Дж. (август 1999 г.). «Молекулярная основа дефицита малонил-КоА-декарбоксилазы» . Американский журнал генетики человека . 65 (2): 318–26. дои : 10.1086/302492 . ПМЦ 1377930 . ПМИД 10417274 .
- ^ Jump up to: а б с Мальвагиа С., Папи Л., Морроне А., Донати М.А., Чиани Ф., Паскини Е., Бренд Дж., Шольте Х.Р., Дженуарди М., Заммарки Е. (ноябрь 2007 г.). «Фатальный дефицит малонил-КоА-декарбоксилазы из-за материнской однородительской изодисомии теломерного конца хромосомы 16». Анналы генетики человека 71 (Часть 6): 705–12. дои : 10.1111/j.1469–1809.2007.00373.x . ПМИД 17535268 . S2CID 35678278 .
- ^ Jump up to: а б с д «Малоновая ацидурия» . Сирота . Проверено 20 апреля 2024 г.
- ^ Браун, ГК; Шолем, Р.Д.; Банкир, А.; Дэнкс, DM (март 1984 г.). «Малонил-коэнзим и дефицит декарбоксилазы» . Журнал наследственных метаболических заболеваний . 7 (1): 21–26. дои : 10.1007/BF01805615 . ISSN 0141-8955 . ПМИД 6145813 .
- ^ Рамасвами, Маматха; Скринска, Виктор Антоний; Абдо, Гассан; Махмуд Ахмед, Лейла; Митри, Рола Файез; Джоши, Рави (март 2017 г.). «Редкий случай малоновой ацидурии, диагностированный при обследовании новорожденных в Катаре» . Международный журнал неонатального скрининга . 3 (1): 5. дои : 10.3390/ijns3010005 . ISSN 2409-515X .
- ^ Девит, М; Деку, я; Вербек, Э; Шот, Р; Шундервурд, Г; Дюран, М; Деклерк, Дж; Хейманс, Дж; Лекин, М; Верхейен, Ф (февраль 2006 г.). «Нарушения головного мозга при дефиците малонил-КоА декарбоксилазы» . Молекулярная генетика и обмен веществ . 87 (2): 102–106. дои : 10.1016/j.ymgme.2005.09.009 . ПМИД 16275149 .
- ^ Jump up to: а б Альфарес, А.; Нуньес, LD; Аль-Тихли, К.; Митчелл, Дж.; Мелансон, С.; Анастасио, Н.; Ха, КЧ; Маевски Дж.; Розенблатт, Д.С.; Браверман, Н. (1 сентября 2011 г.). «Комбинированная малоновая и метилмалоновая ацидурия: секвенирование экзома выявляет мутации гена ACSF3 у пациентов с неклассическим фенотипом» . Журнал медицинской генетики . 48 (9): 602–605. doi : 10.1136/jmedgenet-2011-100230 . ISSN 0022-2593 . ПМИД 21785126 .
- ^ Грегг, Арканзас; Уорман, AW; Торберн, ДР; О'Брайен, МЫ (июнь 1998 г.). «Комбинированная малоновая и метилмалоновая ацидурия с нормальной активностью декарбоксилазы малонил-коэнзима А: случай, подтверждающий множественную этиологию» . Журнал наследственных метаболических заболеваний . 21 (4): 382–390. дои : 10.1023/A:1005302607897 . ISSN 0141-8955 . ПМИД 9700595 .
- ^ МакГарри, Дж. Денис; Браун, Николас Ф. (февраль 1997 г.). «Митохондриальная система карнитинпальмитоилтрансферазы — от концепции к молекулярному анализу» . Европейский журнал биохимии . 244 (1): 1–14. дои : 10.1111/j.1432-1033.1997.00001.x . ISSN 0014-2956 . ПМИД 9063439 .
- ^ Jump up to: а б де Сен-ван дер Фельден, генеральный менеджер Моник; ван дер Хам, Мария; Янс, Джудит Дж.; Рыбак, Гепке; Принц, Хубертус CMT; Верховен-Дуиф, Нанда М.; ван Гассен, Коэн Л.И.; ван Хасселт, Питер М. (2016), Морава, Ева; Баумгартнер, Матиас; Паттерсон, Марк; Рахман, Шамима (ред.), «Новый подход к быстрой метаболической диагностике при CMAMMA», Отчеты JIMD, Том 30 , том. 30, Берлин, Гейдельберг: Springer Berlin Heidelberg, стр. 15–22, номер домена : 10.1007/8904_2016_531 , ISBN. 978-3-662-53680-3 , PMC 5110436 , PMID 26915364
- ^ «КОМБИНИРОВАННАЯ МАЛОНОВАЯ И МЕТИЛМАЛОНОВАЯ АЦИДУРИЯ» . ОМИМ . Проверено 20 апреля 2024 г.
- ^ «Комбинированная малоновая и метилмалоновая ацидемия» . Национальная медицинская библиотека . Проверено 20 апреля 2024 г.
Внешние ссылки
[ редактировать ]- Малоновая ацидурия в NLM Genetics домашнем справочнике