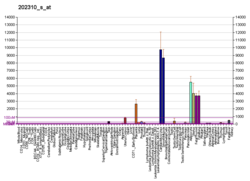
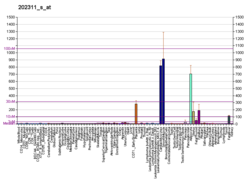
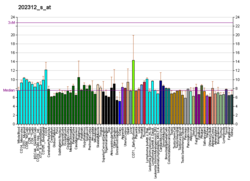
Коллаген - это белок, который укрепляет и поддерживает многие ткани в организме, включая хрящ , кость , сухожилие , кожу и белую часть глаза ( склера ) . Ген COL1A1 производит компонент коллагена типа I , называемый цепью Pro-ALPHA1 (I). Эта цепь в сочетании с другой цепью Pro-ALPHA1 (I), а также с цепью Pro-ALPHA2 (I) (продуцируемой геном COL1A2 ) для создания молекулы проколлагена I типа . Эти тройные, похожие на веревочные молекулы проколлагена должны обрабатываться ферментами вне клетки. После того, как эти молекулы обрабатываются, они заправляются в длинные, тонкие фибрилл, которые сшивают друг друга в пространствах вокруг ячеек. Поперечные связи приводят к формированию очень сильных созревших коллагеновых волокон типа I.
Коллагеновая функция включает в себя жесткость и эластичность.
Мутации в гене COL1A1 связаны со следующими условиями:
Синдром Элеров-Данлос, сосудистый тип : в редких случаях специфические гетерозиготные мутации замены аргинина-цистеина в COL1A1 , которые также связаны с хрупкостью сосудов и имитируют Col3a1-Veds
Синдром Элера -Данлос, тип артрохалазии : он вызван мутациями в гене COL1A1 . Мутации в гене COL1A1 , которые заставляют это расстройство, поручают клетке оставлять часть цепи Pro-ALPHA1 (I), которая содержит сегмент, используемый для прикрепления одной молекулы к другой. Когда эта часть белка отсутствует, структура коллагена I типа подвергается нарушению. Ткани, которые богаты коллагеном I типа, такими как кожа, кости и сухожилия, влияют на это изменение. Ehlers -Danlos тип IV наиболее приписывается аномалиям в ретикулярных волокнах (коллаген тип III).
Синдром Элерса -Данлос, классический тип : в редких случаях было показано, что мутация в гене COL1A1 вызывает синдром классического типа элеров -Данлос. Эта мутация заменяет аминокислотный цистеин для аминокислотного аргинина в положении 134 в белке, изготовленном геном. (Мутация также может быть написана как arg134cys.) Измененный белок взаимодействует ненормально с другими белками-строительством коллагена, нарушая структуру коллагеновых фибриллов I и ловушку коллагена в клетке. Исследователи считают, что эти изменения в коллагене вызывают признаки и симптомы расстройства. Ehlers -Danlos тип IV наиболее приписывается аномалиям в ретикулярных волокнах (коллаген тип III). Без гидроксилирования лизина, ферментом лизил -гидроксилазы, конечная структура коллагена не может образовываться.
Остеогенез несовершенного, тип I : остеогенез несовершенство является наиболее распространенным заболеванием, вызванным мутациями в этом гене. Мутации, которые инактивируют одну из двух копий гена COL1A1 , вызывают остеогенез несовершенного типа I. Мутированная копия гена не вызывает каких-либо коллагеновых цепей Pro-Alpha1 (I). Поскольку только одна копия гена направляет клетку для создания цепей Pro-Alpha1 (i), клетки людей с этим расстройством делают только половину нормального количества коллагена I типа, что приводит к хрупкости кости и другим симптомам.
Остеогенез несовершенного, тип II : Многие различные типы мутаций в гене COL1A1 могут вызвать несовершенство остеогенеза типа II. Эти мутации варьируются от недостающих частей гена COL1A1 до аминокислотных замен, в которых аминокислотный глицин заменяется другой аминокислотой в белковой цепи. Иногда изменяется один конец гена (называемый C-конце), что мешает ассоциации белковых цепей. Все эти изменения предотвращают нормальную выработку созревающего коллагена типа I, что приводит к этому тяжелому состоянию, остеогенезу типа II.
Остеогенез несовершенного, типа III : мутации в гене COL1A1 могут привести к производству белка, в котором отсутствует сегменты, что делает его непригодным для выработки коллагена. Другие мутации приводят к замене аминокислотного глицина другой аминокислотой в цепи проалфа1 (i), которая ингибирует необходимое взаимодействие между белковыми цепями. Производство коллагена типа ингибируется неспособностью измененных цепей проколлагена связывать и сформировать трехцепочечную, надежная структура зрелого коллагена. Эти изменения отрицательно влияют на ткани, богатые коллагеном I типа, такие как кожа, кости, зубы и сухожилия, что приводит к признакам и симптомам остеогенеза III типа.
Остеогенез несовершенного, тип IV : несколько различных типов мутаций в гене Col1a1 вызывают несовершенство остеогенеза типа IV. Эти мутации могут включать отсутствующие части гена COL1A1 или изменения в парах оснований (строительные блоки ДНК). Эти изменения генов приводят к белу, в котором отсутствует сегменты или имеет аминокислотные замены; В частности, аминокислотный глицин заменяется другой аминокислотой. Все эти изменения мешают образованию зрелой молекулы коллагена с тройным цепью и предотвращают производство зрелого коллагена типа I, что приводит к несовершенному остеогенезу типа IV.
Остеопороз : остеопороз - это состояние, которое делает кости постепенно более хрупкими и подверженными разрушению. Особый вариант ( полиморфизм ) в гене COL1A1 , по -видимому, увеличивает риск развития остеопороза . специфическое изменение в сайте связывания SP1 Показано, что связано с повышенным риском низкой костной массы и перелома позвонков из -за изменений, который белок COL1A1 продуцируется из одной копии гена. Несколько исследований показали, что женщины с этой конкретной генетической вариацией в сайте SP1 с большей вероятностью имеют признаки остеопороза, чем женщины без вариации.
Arc.Ask3.Ru Номер скриншота №: 55c2c6c2e61ece16317fb39d67a97d86__1723327140 URL1:https://arc.ask3.ru/arc/aa/55/86/55c2c6c2e61ece16317fb39d67a97d86.html Заголовок, (Title) документа по адресу, URL1: Collagen, type I, alpha 1 - Wikipedia
Данный printscreen веб страницы (снимок веб страницы, скриншот веб страницы), визуально-программная копия документа расположенного по адресу URL1 и сохраненная в файл, имеет: квалифицированную, усовершенствованную (подтверждены: метки времени, валидность сертификата), открепленную ЭЦП (приложена к данному файлу), что может быть использовано для подтверждения содержания и факта существования документа в этот момент времени. Права на данный скриншот принадлежат администрации Ask3.ru, использование в качестве доказательства только с письменного разрешения правообладателя скриншота. Администрация Ask3.ru не несет ответственности за информацию размещенную на данном скриншоте. Права на прочие зарегистрированные элементы любого права, изображенные на снимках принадлежат их владельцам. Качество перевода предоставляется как есть. Любые претензии, иски не могут быть предъявлены. Если вы не согласны с любым пунктом перечисленным выше, вы не можете использовать данный сайт и информация размещенную на нем (сайте/странице), немедленно покиньте данный сайт. В случае нарушения любого пункта перечисленного выше, штраф 55! (Пятьдесят пять факториал, Денежную единицу (имеющую самостоятельную стоимость) можете выбрать самостоятельно, выплаичвается товарами в течение 7 дней с момента нарушения.)