Резонанс (химия)

В химии , резонанс , также называемый мезомерией — это способ описания связей в определенных молекулах или многоатомных ионах путем комбинации нескольких способствующих структур (или форм) . [1] также известные как резонансные структуры или канонические структуры ) в резонансный гибрид (или гибридную структуру ) в теории валентных связей . Это имеет особое значение для анализа делокализованных электронов , связь которых не может быть выражена одной-единственной структурой Льюиса . Резонансный гибрид — это точная структура молекулы или иона; это среднее значение теоретических (или гипотетических) вносящих вклад структур.
Обзор
[ редактировать ]В рамках теории валентных связей резонанс является расширением идеи о том, что связь в химических соединениях может быть описана структурой Льюиса. Для многих химических соединений одна структура Льюиса, состоящая из атомов, подчиняющихся правилу октетов , возможно, несущих формальные заряды и соединенных связями положительного целого порядка, достаточна для описания химической связи и рационализации экспериментально определенных молекулярных свойств, таких как длины связей , углы , и дипольный момент . [2] Однако в некоторых случаях можно нарисовать более одной структуры Льюиса, и экспериментальные свойства несовместимы ни с одной структурой. Чтобы справиться с ситуацией такого типа, несколько вносящих вклад структур рассматриваются вместе как среднее, и говорят, что молекула представляет собой резонансный гибрид, в котором несколько структур Льюиса используются вместе для описания ее истинной структуры.

Например, в № 2 – , нитрит- анион, длины двух связей NO равны, хотя ни одна структура Льюиса не имеет двух связей NO с одинаковым формальным порядком связей . Однако его измеренная структура согласуется с описанием как резонансного гибрида двух основных структур, показанных выше: он имеет две равные связи N–O длиной 125 мкм, промежуточные по длине между типичной одинарной связью N–O (145 мкм в гидроксиламин , H 2 N–OH) и двойная связь N–O (115 пм в ионе нитрония , [O=N=O] + ). В соответствии с участвующими структурами каждая связь NO – O представляет собой среднее значение формальной одинарной и формально двойной связи, что приводит к истинному порядку связи 1,5. Благодаря этому усреднению льюисовское описание связи в NO 2 – согласуется с экспериментальным фактом наличия у аниона эквивалентных связей N–O.
Резонансный гибрид представляет реальную молекулу как «среднее» вносящих вклад структур, при этом длины связей и частичные заряды принимают промежуточные значения по сравнению с ожидаемыми для отдельных структур Льюиса вносящих вклады, если бы они существовали как «реальные» химические образования. . [3] Содействующие структуры различаются только формальным распределением электронов между атомами, а не фактической физически и химически значимой электронной или спиновой плотностью. Хотя вносящие вклад структуры могут различаться формальным порядком связей и формальным распределением заряда , все вносящие вклад структуры должны иметь одинаковое количество валентных электронов и одинаковую спиновую кратность . [4]
Поскольку делокализация электронов снижает потенциальную энергию системы, любая разновидность, представленная резонансным гибридом, более стабильна, чем любая из (гипотетических) вносящих вклад структур. [5] Делокализация электронов стабилизирует молекулу, поскольку электроны более равномерно распределяются по молекуле, уменьшая электрон-электронное отталкивание. [6] Разница в потенциальной энергии между реальными частицами и (вычисленной) энергией вносящей вклад структуры с наименьшей потенциальной энергией называется резонансной энергией. [7] или энергия делокализации. Величина резонансной энергии зависит от предположений, сделанных о гипотетических «нестабилизированных» частицах и используемых вычислительных методах, и не представляет собой измеримую физическую величину, хотя сравнение резонансных энергий, рассчитанных при аналогичных предположениях и условиях, может иметь химический смысл.
Молекулы с расширенной π-системой, такие как линейные полиены и полиароматические соединения, хорошо описываются резонансными гибридами, а также делокализованными орбиталями в теории молекулярных орбиталей .
Резонанс против изомерии
[ редактировать ]Резонанс следует отличать от изомерии . Изомеры — это молекулы с одинаковой химической формулой, но представляющие собой отдельные химические виды с разным расположением атомных ядер в пространстве. С другой стороны, вкладчики резонанса молекулы могут отличаться только тем, как электроны формально приписываются атомам в изображениях структуры Льюиса молекулы. В частности, когда говорят, что молекулярная структура представлена резонансным гибридом, это не означает, что электроны молекулы «резонируют» или перемещаются вперед и назад между несколькими наборами положений, каждое из которых представлено структурой Льюиса. Скорее, это означает, что набор вносящих вклад структур представляет собой промежуточную структуру (средневзвешенное значение вкладчиков) с единой, четко определенной геометрией и распределением электронов. Неверно рассматривать резонансные гибриды как быстро взаимопревращающиеся изомеры, хотя термин «резонанс» может вызвать такой образ. [8] (Как описано ниже , термин «резонанс» возник как аналогия из классической физики квантовомеханического явления, поэтому его не следует понимать слишком буквально.) Символически двунаправленная стрелка используется для обозначения того, что A и B представляют собой формы одного химического вещества (в отличие от стрелки равновесия, например, ; см. ниже подробности об использовании ).


Показательна нехимическая аналогия: характеристики реального животного, нарвала , можно описать с точки зрения характеристик двух мифических существ: единорога , существа с единственным рогом на голове, и левиафана , большого , китообразное существо. Нарвал — это не существо, которое мечется между единорогом и левиафаном, а единорог и левиафан не имеют никакого физического существования за пределами коллективного человеческого воображения. Тем не менее, описание нарвала с точки зрения этих воображаемых существ дает достаточно хорошее описание его физических характеристик.
Из-за путаницы с физическим значением слова «резонанс» , поскольку никакие сущности на самом деле физически не «резонируют», было предложено отказаться от термина «резонанс» в пользу делокализации. [9] и от резонансной энергии отказались в пользу энергии делокализации . Резонансная структура становится содействующей структурой , а резонансный гибрид становится гибридной структурой . Двуглавые стрелки будут заменены запятыми, чтобы проиллюстрировать набор структур, поскольку стрелки любого типа могут указывать на то, что происходит химическое изменение.
Представление на диаграммах
[ редактировать ]![{\displaystyle {\ce {[S=C=N^{\ominus }<->\ ^{\ominus }\!SC{\equiv }N]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/cc41b6c9b3f1ef739335969a581e9a66d5ed0b1a)

На диаграммах участвующие структуры обычно разделяются двунаправленными стрелками (↔). Стрелку не следует путать со стрелками равновесия вправо и влево (⇌). Все структуры вместе могут быть заключены в большие квадратные скобки, чтобы указать, что они изображают одну молекулу или ион, а не разные виды, находящиеся в химическом равновесии .
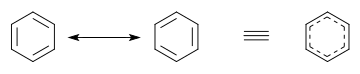
В качестве альтернативы использованию дополнительных структур на диаграммах можно использовать гибридную структуру. В гибридной структуре пи-связи , участвующие в резонансе, обычно изображают в виде кривых [10] или пунктирные линии, указывающие на то, что это частичные, а не нормальные полные пи-связи. В бензоле и других ароматических кольцах делокализованные пи-электроны иногда изображаются в виде сплошного круга. [11]
История
[ редактировать ]Эта концепция впервые появилась в 1899 году в «Гипотезе частичной валентности» Иоганна Тиле , чтобы объяснить необычную стабильность бензола, которую нельзя было ожидать от Августа Кекуле , предложенной в 1865 году с чередующимися одинарными и двойными связями. структуры [12] Бензол вступает в реакции замещения, а не в реакции присоединения, типичные для алкенов . Он предположил, что связь углерод-углерод в бензоле является промежуточной между одинарной и двойной связью.
Предложение о резонансе также помогло объяснить количество изомеров производных бензола. Например, структура Кекуле позволяет предсказать четыре изомера дибромбензола, включая два орто- изомера с бромированными атомами углерода, соединенными одинарной или двойной связью. В действительности существует только три изомера дибромбензола, и только один является орто-изомером, что согласуется с идеей о том, что существует только один тип связи углерод-углерод, промежуточный между одинарной и двойной связью. [13]
Механизм резонанса был введен в квантовую механику Вернером Гейзенбергом в 1926 году при обсуждении квантовых состояний атома гелия. Он сравнил строение атома гелия с классической системой резонирующих связанных гармонических осцилляторов . [3] [14] В классической системе связь порождает две моды, одна из которых по частоте ниже , чем любая из несвязанных колебаний; квантово-механически эта более низкая частота интерпретируется как более низкая энергия. Лайнус Полинг использовал этот механизм для объяснения частичной валентности молекул в 1928 году и развил его дальше в серии статей в 1931–1933 годах. [15] [16] Альтернативный термин мезомерия [17] Популярное в немецких и французских изданиях слово с тем же значением было введено Ч. К. Ингольдом в 1938 году, но не прижилось в английской литературе. Современная концепция мезомерного эффекта приобрела схожий, но другой смысл. Двуглавая стрела была введена немецким химиком Фрицем Арндтом, который предпочитал немецкую фразу zwischenstufe или промежуточную стадию .
Теория резонанса доминировала над конкурирующим методом Хюккеля в течение двух десятилетий благодаря тому, что ее относительно легче понять химикам без фундаментальной физики, даже если они не могли уловить концепцию квантовой суперпозиции и путали ее с таутомерией . охарактеризовали подход Эриха Хюкеля Сами Полинг и Уиланд в то время как «громоздкий», и этому способствовало отсутствие у него коммуникативных навыков: когда Роберт Робинсон отправил ему дружеский запрос, он высокомерно ответил, что органическая химия его не интересует. [18]
В Советском Союзе теория резонанса – особенно разработанная Полингом – подверглась критике в начале 1950-х годов как противоречащая марксистским принципам диалектического материализма , а в июне 1951 года Советская Академия наук под руководством Александра Несмеянова созвала конференцию по химическое строение органических соединений, в котором приняли участие 400 физиков, химиков и философов, где « была раскрыта и разоблачена псевдонаучная сущность теории резонанса». [19]
Основные и второстепенные участники
[ редактировать ]Одна участвующая структура может больше напоминать реальную молекулу, чем другая (в смысле энергии и стабильности). Структуры с низким значением потенциальной энергии более стабильны, чем структуры с высокими значениями, и больше напоминают реальную структуру. Наиболее стабильные вносящие вклад структуры называются основными вкладчиками . Энергетически неблагоприятные и, следовательно, менее выгодные структуры вносят незначительный вклад . Поскольку правила перечислены в примерном порядке по мере убывания важности, основными участниками, как правило, являются структуры, которые
- максимально соблюдать правило октета (8 валентных электронов вокруг каждого атома вместо недостатка или избытка, или 2 электрона для элементов периода 1 );
- иметь максимальное количество ковалентных связей;
- несут минимум формально заряженных атомов , при этом разделение разнородных и одинаковых зарядов минимизировано и максимизировано соответственно;
- поместите отрицательный заряд, если таковой имеется, на наиболее электроотрицательные атомы, а положительный заряд, если таковой имеется, на наиболее электроположительный;
- существенно не отклоняться от идеализированных длин связей и углов (например, относительная незначительность вкладчиков резонанса типа Дьюара для бензола);
- локально сохранять ароматические субструктуры, избегая при этом антиароматических ( см. секстет Клара и бифенилен ).
Максимум восемь валентных электронов является строгим для элементов Периода 2 Be, B, C, N, O и F, как и максимум два для H и He, а также эффективно для Li. [20] Спорным является вопрос о расширении валентной оболочки третьего периода и более тяжелых элементов основной группы. Структура Льюиса, в которой центральный атом имеет число валентных электронов больше восьми, традиционно предполагает участие d-орбиталей в связывании. Однако общепринятое мнение состоит в том, что, хотя они могут вносить незначительный вклад, участие d-орбиталей не имеет значения, а связывание так называемых гипервалентных молекул по большей части лучше объясняется вкладывающими формами с разделенными зарядами, которые изображают три -центральная четырехэлектронная связь . Тем не менее, по традиции, структуры расширенных октетов по-прежнему обычно рисуются для таких функциональных групп, как, например, сульфоксиды , сульфоны и илиды фосфора . Рассматриваемые как формализм, который не обязательно отражает истинную электронную структуру, IUPAC предпочитает такие изображения структурам с частичными связями, разделением зарядов или дативными связями . [21]
Эквивалентные участники вносят одинаковый вклад в фактическую структуру, тогда как важность неэквивалентных участников определяется степенью их соответствия перечисленным выше свойствам. Большее количество значимых структур и более объемное пространство, доступное для делокализованных электронов, приводят к стабилизации (понижению энергии) молекулы.
Примеры
[ редактировать ]Ароматические молекулы
[ редактировать ]В бензоле две циклогексатриеновые структуры Кекуле , впервые предложенные Кекуле , взяты вместе как способствующие структуры, чтобы представить общую структуру. В гибридной структуре справа пунктирный шестиугольник заменяет три двойные связи и представляет шесть электронов в наборе из трех молекулярных орбиталей симметрии π- с узловой плоскостью в плоскости молекулы.
В фуране неподеленная пара атома кислорода взаимодействует с π-орбиталями атомов углерода. Изогнутые стрелки изображают перестановку делокализованных π-электронов , в результате которой вносят разные вклады.

Молекулы, богатые электронами
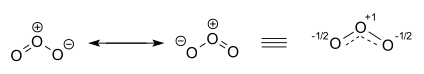
[ редактировать ]Молекула озона представлена двумя участвующими структурами. На самом деле два концевых атома кислорода эквивалентны, и гибридная структура изображена справа с зарядом — 1 ⁄ 2 как на атомах кислорода, так и на частичных двойных связях с полной и пунктирной линией и порядком связи. 1 + 1 ⁄ 2 . [22] [23]
Для гипервалентных молекул описанная выше рационализация может быть применена для создания структур, способствующих объяснению связей в таких молекулах. Ниже показаны структуры, способствующие образованию связи 3c-4e в дифториде ксенона .
![{\displaystyle {\ce {[{\mathsf {F-XeF^{-}<->F^{-}Xe-F}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6841f8221ad24ec2c691c3280284c43996ad1ffb)
Электронодефицитные молекулы
[ редактировать ]Аллильный катион имеет две способствующие структуры с положительным зарядом на концевых атомах углерода. В гибридной структуре их заряд + 1 ⁄ 2 . Полный положительный заряд также можно изобразить как делокализованный между тремя атомами углерода.

Молекула диборана описывается участвующими структурами, каждая из которых имеет дефицит электронов на разных атомах. Это уменьшает дефицит электронов на каждом атоме и стабилизирует молекулу. Ниже приведены структуры, способствующие отдельной связи 3c-2e в диборане.
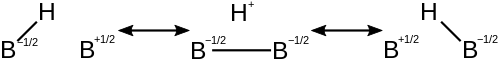
Реактивные промежуточные продукты
[ редактировать ]Часто реакционноспособные промежуточные соединения, такие как карбокатионы и свободные радикалы, имеют более делокализованную структуру, чем их исходные реагенты, что приводит к образованию неожиданных продуктов. Классический пример — аллильная перегруппировка . [24] Когда 1 моль HCl присоединяется к 1 молю 1,3-бутадиена, помимо обычно ожидаемого продукта 3-хлор-1-бутена мы также обнаруживаем 1-хлор-2-бутен. Эксперименты по мечению изотопов показали, что здесь происходит то, что дополнительная двойная связь смещается из положения 1,2 в положение 2,3 в некоторых продуктах. Это и другие данные (например, ЯМР в растворах суперкислот ) показывают, что промежуточный карбокатион должен иметь сильно делокализованную структуру, отличную от его в основном классической (делокализация существует, но невелика) родительской молекулы. Этот катион (аллильный катион) можно представить с помощью резонанса, как показано выше.
Это наблюдение большей делокализации в менее стабильных молекулах является весьма общим. Возбужденные состояния сопряженных диенов стабилизируются в большей степени за счет сопряжения, чем их основные состояния, в результате чего они становятся органическими красителями. [25]
Хорошо изученный пример делокализации без участия π-электронов ( гиперконъюгация ) можно наблюдать в неклассическом 2-норборнильном катионе. [26] Другой пример - метан ( CH +
5 ). Их можно рассматривать как содержащие трехцентровые двухэлектронные связи и они представлены либо вносящими вклад структурами, включающими перегруппировку σ-электронов, либо специальным обозначением Y, в трех точках которого находятся три ядра.
Делокализованные электроны важны по нескольким причинам; Главный из них заключается в том, что ожидаемая химическая реакция может не произойти, поскольку электроны делокализуются в более стабильную конфигурацию, в результате чего реакция происходит в другом месте. Примером является Фриделя-Крафтса. алкилирование [27] бензола с 1-хлор-2-метилпропаном; карбокатион группу , перегруппировывается в трет - бутильную стабилизированную гиперконъюгацией , особой формой делокализации.
Бензол
[ редактировать ]Длина связи
[ редактировать ]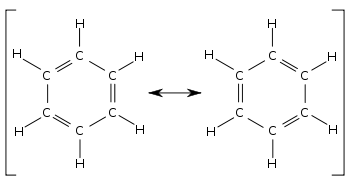
При сравнении двух участвующих структур бензола все одинарные и двойные связи меняются местами. Длины связей можно измерить, например, с помощью дифракции рентгеновских лучей . Средняя длина одинарной связи C–C составляет 154 пм ; для двойной связи C=C — 133 пм. В локализованном циклогексатриене связи углерод-углерод должны чередоваться 154 и 133 пм. Вместо этого обнаружено, что все связи углерод-углерод в бензоле имеют длину около 139 пм, что является промежуточной длиной связи между одинарной и двойной связью. Этот смешанный характер одинарных и двойных (или тройных) связей типичен для всех молекул, в которых связи имеют разный порядок связей в разных участвующих структурах. Длину облигаций можно сравнить с помощью ордеров на облигации. Например, в циклогексане порядок связи равен 1, а в бензоле – 1 + (3 ÷ 6) = 1 + 1 ⁄ 2 . Следовательно, бензол имеет более двойную связь и, следовательно, более короткую длину связи, чем циклогексан.
Резонансная энергия
[ редактировать ]Энергия резонанса (или делокализации) — это количество энергии, необходимое для преобразования истинной делокализованной структуры в структуру наиболее стабильной способствующей структуры. Эмпирическую резонансную энергию можно оценить путем сравнения изменения энтальпии реального гидрирования вещества с оценкой для вносящей вклад структуры.
Полное гидрирование бензола до циклогексана через 1,3-циклогексадиен и циклогексен является экзотермическим ; 1 моль бензола дает 208,4 кДж (49,8 ккал).

Гидрирование одного моля двойных связей дает 119,7 кДж (28,6 ккал), как следует из последнего этапа - гидрирования циклогексена. Однако в бензоле для гидрирования одного моля двойных связей необходимо 23,4 кДж (5,6 ккал). Разница, составляющая 143,1 кДж (34,2 ккал), представляет собой эмпирическую резонансную энергию бензола. Поскольку 1,3-циклогексадиен также имеет небольшую энергию делокализации (7,6 кДж или 1,8 ккал/моль), чистая резонансная энергия по сравнению с локализованным циклогексатриеном немного выше: 151 кДж или 36 ккал/моль. [28]
Эта измеренная резонансная энергия также представляет собой разницу между энергией гидрирования трех «нерезонансных» двойных связей и измеренной энергией гидрирования:
- (3 × 119,7) − 208,4 = 150,7 кДж/моль (36 ккал). [29]
Независимо от их точных значений, резонансные энергии различных родственных соединений дают представление об их связи. Резонансные энергии для пиррола , тиофена и фурана составляют соответственно 88, 121 и67 кДж/моль (21, 29 и 16 ккал/моль). [30] Таким образом, эти гетероциклы гораздо менее ароматичны, чем бензол, что проявляется в лабильности этих колец.
Квантово-механическое описание в теории валентной связи (ВС)
[ редактировать ]
Резонанс имеет более глубокое значение в математическом аппарате теории валентных связей (ВС). Квантовая механика требует, чтобы волновая функция молекулы подчинялась наблюдаемой симметрии. Если какая-то одна структура не достигает этого, возникает резонанс.
Например, в бензоле теория валентных связей начинается с двух структур Кекуле, которые по отдельности не обладают шестикратной симметрией реальной молекулы. Теория строит реальную волновую функцию как линейную суперпозицию волновых функций, представляющих две структуры. Поскольку обе структуры Кекуле имеют одинаковую энергию, они вносят одинаковый вклад в общую структуру - суперпозиция представляет собой равновзвешенное среднее или линейную комбинацию этих двух в соотношении 1:1 в случае бензола. Как показано , симметричная комбинация дает основное состояние, а антисимметричная комбинация дает первое возбужденное состояние .
Обычно суперпозиция записывается с неопределенными коэффициентами, которые затем вариационно оптимизируются , чтобы найти минимально возможную энергию для данного набора базисных волновых функций. Когда включено больше вносящих вклад структур, молекулярная волновая функция становится более точной, и больше возбужденных состояний можно получить из различных комбинаций вносящих вклад структур.
Сравнение с теорией молекулярных орбиталей (МО)
[ редактировать ]
В теории молекулярных орбиталей , основной альтернативе теории валентных связей , молекулярные орбитали (МО) аппроксимируются как суммы всех атомных орбиталей (АО) на всех атомах; МО столько же, сколько и АО. Каждый АО i имеет весовой коэффициент c i , который указывает вклад АО в конкретную МО. Например, в бензоле модель МО дает нам 6 π-МО, которые представляют собой комбинации 2p z АО на каждом из 6 атомов C. Таким образом, каждая π-МО делокализована по всей молекуле бензола, а любой электрон, занимающий МО, будет делокализован по всей молекуле. Эта интерпретация МО вдохновила представление о бензольном кольце как шестиугольнике с кругом внутри. При описании бензола в курсах элементарной химии часто объединяют концепцию ВБ о локализованных σ-связях и концепцию МО о делокализованных π-орбиталях.
Структуры, способствующие модели VB, особенно полезны для прогнозирования влияния заместителей на π-системы, такие как бензол. Они приводят к моделям способствующих структур электроноакцепторной группы и электроновысвобождающей группы бензола. Полезность теории МО заключается в том, что количественное указание заряда π-системы на атоме можно получить из квадратов весового коэффициента c i на атоме C i . Заряд q i ≈ c 2
я . Причина возведения коэффициента в квадрат заключается в том, что если электрон описывается АО, то квадрат АО дает плотность электронов . АО корректируются ( нормализуются ) так, что АО 2 знак равно 1, и q я ≈ ( c я AO i ) 2 ≈ с 2
я . В бензоле q i = 1 на каждом атоме углерода. С электроноакцепторной группой q i < 1 на орто- и пара- атомах С и q i > 1 для электроно-отдающей группы .
Коэффициенты
[ редактировать ]Взвешивание участвующих структур с точки зрения их вклада в общую структуру можно рассчитать несколькими способами, используя методы «Ab initio» , полученные из теории валентных связей, или же из подходов орбиталей естественных связей (NBO) Weinhold NBO5. Архивировано 2008 г. 02-08 на Wayback Machine или, наконец, на основе эмпирических расчетов на основе метода Хюккеля. Программное обеспечение для обучения резонансу, основанное на методе Хюккеля, доступно на веб-сайте HuLiS .
Делокализация заряда
[ редактировать ]В случае ионов принято говорить о делокализованном заряде (делокализации заряда). Пример делокализованного заряда ионов можно найти в карбоксилатной группе, в которой отрицательный заряд одинаково сосредоточен на двух атомах кислорода. Делокализация заряда анионов является важным фактором, определяющим их реакционную способность (обычно: чем выше степень делокализации, тем ниже реакционная способность) и, в частности, кислотную силу сопряженных с ними кислот. Как правило, чем лучше делокализован заряд аниона, тем сильнее его сопряженная кислота . Например, отрицательный заряд перхлорат- аниона ( ClO −
4 ) равномерно распределен среди симметрично ориентированных атомов кислорода (причем часть его удерживается и центральным атомом хлора). Эта превосходная делокализация заряда в сочетании с большим количеством атомов кислорода (четыре) и высокой электроотрицательностью центрального атома хлора приводит к тому, что хлорная кислота является одной из самых сильных известных кислот со значением ap -10 K . [32] Степень делокализации заряда в анионе может быть количественно выражена через параметр WAPS (средневзвешенная положительная сигма). [33] параметр и аналогичный WANS (средневзвешенная отрицательная сигма) [34] [35] параметр используется для катионов.
| Сложный | ВАПС × 10 5 | Сложный | ВАНС × 10 5 |
|---|---|---|---|
| (C 2 F 5 SO 2 ) 2 NH | 2.0 [36] | Трифенилфосфин | 2.1 [34] |
| (CF 3 ) 3 COH | 3.6 [36] | Фенилтетраметилгуанидин | 2.5 [34] |
| Пикриновая кислота | 4.3 [33] | Трипропиламин | 2.6 [34] |
| 2,4-динитрофенол | 4.9 [33] | МТБД ( 7-метилтриазабициклодецен ) | 2.9 [35] |
| Бензойная кислота | 7.1 [33] | ДБУ ( 1,8-диазабициклоундек-7-ен ) | 3.0 [35] |
| Фенол | 8.8 [36] | TBD ( Триазабициклодецен ) | 3.5 [35] |
| Уксусная кислота | 16.1 [33] | N , N -Диметиланилин | 4.7 [34] |
| ПРИВЕТ | 21.9 [36] | Пиридин | 7.2 [34] |
| ХБр | 29.1 [36] | Анилин | 8.2 [34] |
| HCl | 35.9 [33] | Пропиламин | 8.9 [34] |
Значения WAPS и WANS указаны в е / Å. 4 . Большие значения указывают на более локализованный заряд соответствующего иона.
См. также
[ редактировать ]- Теория молекулярных орбиталей Хюккеля
- Сопряженная система
- Флюксиальная молекула
- Избегали пересечения
Внешние ссылки
[ редактировать ]- Гудар, Н.; Кариссан, Ю.; Хагебаум-Ренье, Д.; Хумбель, С. (2008). «HuLiS: Java-апплет — простая теория Хюккеля и мезомерия — программное обеспечение логики программы» (на французском языке) . Проверено 29 октября 2010 г.
Ссылки
[ редактировать ]- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Онлайн исправленная версия: (2006–) « Резонанс ». дои : 10.1351/goldbook.R05326
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) « Содействующая структура ». дои : 10.1351/goldbook.C01309
- ^ Jump up to: а б Полинг, Лайнус (1960). «Понятие резонанса» . Природа химической связи - Введение в современную структурную химию (3-е изд.). Издательство Корнельского университета. стр. 10–13. ISBN 978-0801403330 .
- ^ Практикующие химики, знакомые с концепциями резонанса и делокализации, часто рисуют только одну основную структуру, которая неявно представляет молекулу, структуру которой следует описать, используя резонансный гибрид. Например, химик может произвольно нарисовать резонансный вклад NO 2 – показано слева, с учетом того, что читатель знает о другом вкладчике, показанном справа, а также подразумевается, что связи NO на самом деле эквивалентны. Эта практика особенно распространена в органической химии, где одна из структур Кекуле бензола часто выбирается для изображения правильной гексагональной структуры молекулы.
- ^ Моррисон, Роберт; Бойд, Роберт (1989). «Глава 10». Органическая химия (5-е изд.). Прентис Холл Индии. п. 372. ИСБН 978-0-87692-560-7 .
Резонансный гибрид более стабилен, чем любая из участвующих структур.
- ^ Кэри, Фрэнсис А.; Сундберг, Ричард Дж. (2007). Продвинутая органическая химия. Часть A: Структура и механизмы . Спрингер. п. 19. ISBN 978-0-387-68346-1 .
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Онлайн исправленная версия: (2006–) « Энергия резонанса ». дои : 10.1351/goldbook.R05333
- ^ «Резонансные формы» . UCDavis Chem Wiki . UCDavis. 02.10.2013 . Проверено 7 октября 2015 г.
- ^ Кербер, Роберт С. (2006). «Если это резонанс, то что резонирует?». Дж. Хим. Образование . 83 (2): 223. Бибкод : 2006ЖЧЭд..83..223К . дои : 10.1021/ed083p223 .
- ^ «Графическое представление диаграмм химической структуры» (PDF) , Рекомендации ИЮПАК 2008 г. , ИЮПАК , стр. 387 (ГР–8)
- ^ «Графическое представление диаграмм химической структуры» (PDF) , Рекомендации ИЮПАК 2008 г. , ИЮПАК , стр. 379–382 (GR–6)
- ^ Тиле, Йоханнес (1899). «Zur Kenntnis der ungesättigten Verbindungen» [[Вклад] в наши знания о ненасыщенных соединениях]. Annalen der Chemie Юстуса Либиха (на немецком языке). 306 : 87–142. дои : 10.1002/jlac.18993060107 . На стр. 89, Тиле ввел понятие «частичной валентности»: «Ich nehme nun an, ... eine Partialvalens vorhanden ist, eine Annahme, die sich auch thermisch begründen lässt». (Теперь я предполагаю, что в случае веществ, которым приписана двойная связь, фактически для их связи используются два сродства каждого из участвующих атомов; однако из-за способности к присоединению двойных связей сила сродства не полностью израсходован, и в каждом из атомов существует остаток сродства или «частичная валентность» — предположение, которое можно обосновать и термически (т. е. с помощью калориметрии).) На с. 90, Тиле ввел термин «сопряженный»: «Ein solches System benachbarter Doppelbindungen mit ausgeglichenen Insiden Partialvalenzen sei als conjugirt bezeichnet». (Такую систему соседних двойных связей с уравненными внутренними частичными валентностями следует называть «сопряженной».) Тиле обсуждал сопряженную структуру бензола на стр. 125–129: VIII. Die Aromatischen Verbindungen. Дас Бензол. (VIII. Ароматические соединения. Бензол.)
- ^ Хорнбек, Джозеф М. (2006). Органическая химия (2-е изд.). Томсон Обучение. стр. 470–1. ISBN 9780534389512 .
- ^ Полинг, Лайнус, Резонанс , с. 1
- ^ «Наука и гуманизм Лайнуса Полинга» . Архивировано из оригинала 31 марта 2012 г.
{{cite web}}: CS1 maint: bot: статус исходного URL неизвестен ( ссылка ). См. последний абзац раздела 1. - ^ Полинг, Л. (1960). Природа химической связи (3-е изд.). Издательство Оксфордского университета. п. 184 . В этом источнике Полинг сначала упоминает соответствующие работы Слейтера и Хюкеля в 1931 году, а затем цитирует свои собственные ключевые статьи: Полинг, Лайнус. (1931). «Природа химической связи. Ii. Одноэлектронная связь и трехэлектронная связь». Дж. Ам. хим. Соц . 53 (1367): 3225. doi : 10.1021/ja01360a004 . и последующие статьи 1932–33 гг.
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Интернет-исправленная версия: (2006–) « Мезомерия ». два : 10.1351/goldbook.M03845
- ^ Моррис, Питер Дж.Т.; Хорникс, Уиллем Дж.; Бад, Роберт; Моррис, Питер Дж. Т. (1992). «Технология: взаимодействие науки: Уолтер Реппе и химия циклооктатетраена» . Британский журнал истории науки . 25 (1): 145–167. дои : 10.1017/S0007087400045374 . JSTOR 4027009 . S2CID 145124799 .
- ^ Мур, Баррингтон младший (1954). Террор и прогресс СССР: некоторые источники перемен и стабильности советской диктатуры . стр. 142–143.
- ^ Литий всегда встречается как Li + (1 с 2 ), дуэт, в ионных соединениях. В соединениях типа CH 3 Li с некоторой степенью ковалентности связь достигается преимущественно за счет 2s-орбитали с некоторым вкладом 2p-орбитали. (Эта схема связи также используется в агрегатах конденсированной фазы, таких как (CH 3 Li) 4 , что приводит к более высокому координационному числу лития.) Таким образом, в принципе можно разместить до октета. Тем не менее формальное число валентных электронов вокруг Li никогда не превышает двух, если только не учитывать слабые донорно-акцепторные взаимодействия с нейтральными лигандами (например, молекулами растворителя, которые часто исключаются из структур Льюиса).
- ^ Бречер, Джонатан (1 января 2008 г.). «Стандарты графического представления диаграмм химической структуры (Рекомендации ИЮПАК 2008 г.)» . Чистая и прикладная химия . 80 (2): 277–410. дои : 10.1351/pac200880020277 . ISSN 1365-3075 .
- ^ Уэйд, Г. Органическая химия (6-е изд.). [ ISBN отсутствует ]
- ^ Брюс, Паула Ю. Органическая химия (4-е изд.). [ ISBN отсутствует ]
- ^ Эшенхерст, Джеймс (2 декабря 2013 г.). «Бонусная тема: Аллильные перегруппировки» . Магистр органической химии . Проверено 7 февраля 2024 г.
- ^ «16.12 Сопряженные диены и УФ-свет» . Химия LibreTexts . 01.04.2015 . Проверено 7 февраля 2024 г.
- ^ Мосс, Роберт А. (4 февраля 2014 г.). «2-норборнильный катион: ретроспектива» . Журнал физической органической химии . 27 (5): 374–379. дои : 10.1002/poc.3290 . ISSN 0894-3230 - через онлайн-библиотеку Wiley.
- ^ «4.10: Алкилирование и ацилирование ароматических колец - реакция Фриделя-Крафтса» . Химия LibreTexts . 21 июня 2020 г. Проверено 7 февраля 2024 г.
- ^ Виберг; Накадзи; Морган (1993). «Теплость гидрирования цисимина . Экспериментальное и теоретическое исследование». Дж. Ам. хим. Соц . 115 (9): 3527–3532. дои : 10.1021/ja00062a017 .
- ^ Шерман, Дж. (февраль 1939 г.). «Теплоты гидрирования непредельных углеводородов» . Дж. Ам. Нефть Хим. Соц . 16 (2): 28. дои : 10.1007/BF02543208 . S2CID 96029597 . Архивировано из оригинала 14 июля 2011 г.
- ^ Смит, Майкл Б.; Марч, Джерри (2007), Продвинутая органическая химия: реакции, механизмы и структура (6-е изд.), Нью-Йорк: Wiley-Interscience, стр. 62, ISBN 978-0-471-72091-1
- ^ Шайк, Сасон С.; Хиберти, Филипп К. (2008). Руководство химика по теории валентных связей . Нью-Джерси: Wiley-Interscience. стр. 200–203 . ISBN 978-0-470-03735-5 .
- ^ Селлерс, Кэтлин; Уикс, Кэтрин; Олсоп, Уильям Р.; Клаф, Стивен Р.; Хойт, Мэрилин; Пью, Барбара (2006). Перхлорат: Экологические проблемы и решения . ЦРК Пресс. п. 16. ISBN 978-0-8493-8081-5 .
- ^ Jump up to: а б с д и ж Каупмеес, К.; Кальюранд, И.; Лейто, И. (2010). «Влияние содержания воды на кислотность в ацетонитриле. Количественная оценка делокализации заряда в анионах». Дж. Физ. хим. А. 114 (43): 11788–11793. Бибкод : 2010JPCA..11411788K . дои : 10.1021/jp105670t . ПМИД 20919704 .
- ^ Jump up to: а б с д и ж г час Каупмеес, К.; Кальюранд, И.; Лейто, И. (2014). «Влияние содержания воды на основность ацетонитрила». Дж. Солют. Хим . 43 (7): 1270–1281. дои : 10.1007/s10953-014-0201-4 . S2CID 95538780 .
- ^ Jump up to: а б с д Каупмеес, К.; Труммал, А.; Лейто, И. (2014). «Основы сильных оснований в воде: вычислительное исследование» . Хорват. хим. Акта . 87 (4): 385–395. дои : 10.5562/cca2472 .
- ^ Jump up to: а б с д и Книга, Э.; Каупмеес, К.; Овсянников Г.; Драм, А.; Хантер, А.; Сааме, Дж.; Коппель, И.; Кальюранд, И.; Липпинг, Л.; Родима, Т.; Пиль, В.; Коппель, Айова; Лейто, И. (2013). «Кислотность сильных нейтральных кислот Бренстеда в разных средах». Дж. Физ. Орг. Хим . 26 (2): 162–170. дои : 10.1002/poc.2946 .
| Базы данных органов управления : Национальные |
|---|