Экспрессия GALT контролируется действием гена FOXO3 . Отсутствие этого фермента приводит к классической галактоземии у человека и может привести к летальному исходу в период новорожденности, если не исключить лактозу из рациона. Патофизиология галактоземии четко не определена. [5]
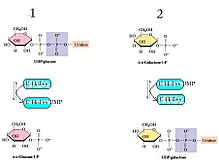
GALT катализирует вторую реакцию пути метаболизма галактозы Лелуара посредством кинетики «пинг-понг би-би» с механизмом двойного замещения . [6] Это означает, что конечная реакция состоит из двух реагентов и двух продуктов (см. реакцию выше) и протекает по следующему механизму: фермент реагирует с одним субстратом с образованием одного продукта и модифицированного фермента, который далее реагирует со вторым субстратом. субстрат для получения второго продукта при регенерации исходного фермента. [7] В случае GALT остаток His166 действует как мощный нуклеофил, облегчая перенос нуклеотида между УДФ-гексозами и гексозо-1-фосфатами. [8]
Трехмерная структура с 180 пм разрешением ( рентгеновская кристаллография ) GALT была определена Ведекиндом, Фреем и Рэйментом, и их структурный анализ обнаружил ключевые аминокислоты, необходимые для функции GALT. [8] Среди них Leu4, Phe75, Asn77, Asp78, Phe79 и Val108, которые соответствуют остаткам, которые участвовали как в экспериментах по точечным мутациям, так и в клиническом скрининге, которые играют роль в галактоземии человека. [8] [10]
GALT также имеет минимальную (~0,1%) активность трансферазы GalNAc. Рентгеновская кристаллография показала, что боковая цепь Tyr289 образует водородную связь с N -ацетильной группой UDP-GalNAc . Точечная мутация остатка Tyr289 на Leu, Ile или Asn устраняет это взаимодействие, повышая активность трансферазы GalNAc, при этом мутация Y289L демонстрирует сопоставимую активность трансферазы GalNAc с активностью трансферазы Gal фермента дикого типа. [11]
Дефицит GALT вызывает классическую галактоземию . Галактоземия — аутосомно-рецессивное наследственное заболевание, выявляемое у новорожденных и детей раннего возраста. [12] Встречается примерно у 1 из каждых 40 000–60 000 живорожденных детей. Классическая галактоземия (G/G) вызвана дефицитом активности GALT, тогда как более распространенные клинические проявления Дуарте (D/D) и вариант Дуарте/классический (D/G) вызваны ослаблением активности GALT. [13] Симптомы включают недостаточность яичников, нарушение координации развития (трудности говорить правильно и последовательно), [14] и неврологический дефицит. [13] Единственная мутация в любой из нескольких пар оснований может привести к дефициту активности GALT. [15] Например, единственная мутация от A до G в экзоне 6 гена GALT превращает Glu188 в аргинин , а мутация от A до G в экзоне 10 превращает Asn314 в аспарагиновую кислоту . [13] Эти две мутации также добавляют новые сайты разрезания ферментов рестрикции , которые позволяют обнаруживать их с помощью широкомасштабного популяционного скрининга с помощью ПЦР ( полимеразная цепная реакция ). [13] Скрининг в основном исключил неонатальную смертность от G/G-галактоземии, но это заболевание, из-за роли GALT в биохимическом метаболизме потребляемой галактозы (которая при накоплении токсична) в энергетически полезную глюкозу , безусловно, может быть фатальным. [12] [16] Тем не менее, люди, страдающие галактоземией, могут жить относительно нормальной жизнью, избегая молочных продуктов и всего, что содержит галактозу (поскольку она не может метаболизироваться), но все же существует вероятность проблем с неврологическим развитием или других осложнений, даже у тех, кто избегает галактозы. [17]
Райхардт Дж. К., Леви Х. Л., Ву С. Л. (июнь 1992 г.). «Молекулярная характеристика двух мутаций галактоземии и одного полиморфизма: значение для структурно-функционального анализа галактозо-1-фосфатуридилтрансферазы человека». Биохимия . 31 (24): 5430–3. дои : 10.1021/bi00139a002 . ПМИД 1610789 .
Флах Дж. Э., Райхардт Дж. К., Эльзас Л. Дж. (август 1990 г.). «Последовательность кДНК, кодирующей галактозо-1-фосфатуридилтрансферазу человека». Молекулярная биология и медицина . 7 (4): 365–9. ПМИД 2233247 .
Райхардт Дж. К., Берг П. (апрель 1988 г.). «Клонирование и характеристика кДНК, кодирующей галактозо-1-фосфатуридилтрансферазу человека». Молекулярная биология и медицина . 5 (2): 107–22. ПМИД 2840550 .
Arc.Ask3.Ru Номер скриншота №: b5463280ec58210c539fc5e70816e746__1703023560 URL1:https://arc.ask3.ru/arc/aa/b5/46/b5463280ec58210c539fc5e70816e746.html Заголовок, (Title) документа по адресу, URL1: Galactose-1-phosphate uridylyltransferase - Wikipedia
Данный printscreen веб страницы (снимок веб страницы, скриншот веб страницы), визуально-программная копия документа расположенного по адресу URL1 и сохраненная в файл, имеет: квалифицированную, усовершенствованную (подтверждены: метки времени, валидность сертификата), открепленную ЭЦП (приложена к данному файлу), что может быть использовано для подтверждения содержания и факта существования документа в этот момент времени. Права на данный скриншот принадлежат администрации Ask3.ru, использование в качестве доказательства только с письменного разрешения правообладателя скриншота. Администрация Ask3.ru не несет ответственности за информацию размещенную на данном скриншоте. Права на прочие зарегистрированные элементы любого права, изображенные на снимках принадлежат их владельцам. Качество перевода предоставляется как есть. Любые претензии, иски не могут быть предъявлены. Если вы не согласны с любым пунктом перечисленным выше, вы не можете использовать данный сайт и информация размещенную на нем (сайте/странице), немедленно покиньте данный сайт. В случае нарушения любого пункта перечисленного выше, штраф 55! (Пятьдесят пять факториал, Денежную единицу (имеющую самостоятельную стоимость) можете выбрать самостоятельно, выплаичвается товарами в течение 7 дней с момента нарушения.)