Эпигенетика рака
Эпигенетика рака — это исследование эпигенетических модификаций ДНК раковых клеток , которые не предполагают изменения нуклеотидной последовательности, а вместо этого включают изменение способа выражения генетического кода. Эпигенетические механизмы необходимы для поддержания нормальных последовательностей экспрессии тканеспецифичных генов и имеют решающее значение для нормального развития. [1] Они могут быть столь же важными, если не даже более важными, чем генетические мутации в трансформации клетки в рак. Нарушение эпигенетических процессов при раке может привести к потере экспрессии генов , что происходит примерно в 10 раз чаще вследствие подавления транскрипции (вызванного гиперметилированием эпигенетического промотора CpG-островков ), чем мутаций. Как Фогельштейн и др. указывает, что при колоректальном раке обычно имеется от 3 до 6 мутаций водителя и от 33 до 66 мутаций автостопщика или пассажира. [2] Однако в опухолях толстой кишки по сравнению с прилегающей нормальной на вид слизистой оболочкой толстой кишки в промоторах генов опухолей имеется от 600 до 800 сильно метилированных CpG-островков, тогда как в прилегающей слизистой оболочке эти CpG-островки не метилированы. [3] [4] [5] Манипулирование эпигенетическими изменениями открывает большие перспективы для профилактики, выявления и терапии рака. [6] [7] рака могут быть нарушены различные эпигенетические механизмы, такие как подавление генов-супрессоров опухоли и активация онкогенов При различных типах за счет изменения CpG-островков паттернов метилирования , модификаций гистонов и нарушения регуляции ДНК-связывающих белков . Существует несколько препаратов, оказывающих эпигенетическое воздействие, которые в настоящее время используются при ряде этих заболеваний.


Механизмы
[ редактировать ]Метилирование ДНК
[ редактировать ]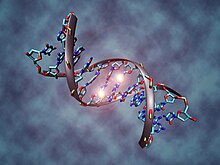
В соматических клетках закономерности метилирования ДНК обычно передаются дочерним клеткам с высокой точностью. [8] Обычно это метилирование происходит только в цитозинах, которые расположены с 5'-конца по отношению к гуанозину в динуклеотидах CpG эукариот более высокого порядка. [9] Однако эпигенетическое метилирование ДНК различается в нормальных и опухолевых клетках человека. «Нормальный» профиль метилирования CpG часто инвертируется в клетках, которые становятся онкогенными. [10] В нормальных клетках островки CpG , предшествующие промоторам генов , обычно неметилированы и имеют тенденцию быть транскрипционно активными, в то время как другие отдельные динуклеотиды CpG по всему геному имеют тенденцию быть метилированными. Однако в раковых клетках островки CpG , предшествующие промоторам генов-супрессоров опухоли , часто гиперметилированы, тогда как метилирование CpG областей промотора онкогена и последовательностей паразитных повторов часто снижено. [11]
Гиперметилирование областей промотора гена-супрессора опухоли может привести к молчанию этих генов. Этот тип эпигенетической мутации позволяет клеткам бесконтрольно расти и размножаться, что приводит к онкогенезу. [10] Добавление метильных групп к цитозинам приводит к тому, что ДНК плотно скручивается вокруг белков-гистонов, в результате чего ДНК не может подвергаться транскрипции (ДНК с молчанием транскрипции). Гены, которые обычно подавляют транскрипцию из-за гиперметилирования промотора, включают: ингибитор циклин-зависимой киназы p16 , ингибитор клеточного цикла; MGMT , ген репарации ДНК ; APC – регулятор клеточного цикла; MLH1 , ген репарации ДНК; и BRCA1 , еще один ген репарации ДНК. [10] [12] Действительно, раковые клетки могут стать зависимыми от подавления транскрипции из-за гиперметилирования промотора некоторых ключевых генов-супрессоров опухоли — процесс, известный как эпигенетическая зависимость. [13]
Гипометилирование динуклеотидов CpG в других частях генома приводит к нестабильности хромосом из-за таких механизмов, как потеря импринтинга и реактивация мобильных элементов . [14] [15] [16] [17] Потеря импринтинга гена инсулиноподобного фактора роста (IGF2) увеличивает риск колоректального рака и связана с синдромом Беквита-Видемана , который значительно увеличивает риск рака у новорожденных. [18] В здоровых клетках CpG-динуклеотиды более низких плотностей обнаруживаются в кодирующих и некодирующих межгенных областях. Экспрессия некоторых повторяющихся последовательностей и мейотическая рекомбинация на центромерах подавляются посредством метилирования. [19]
Весь геном раковой клетки содержит значительно меньше метилцитозина , чем геном здоровой клетки. Фактически, геномы раковых клеток имеют на 20-50% меньше метилирования отдельных динуклеотидов CpG по всему геному. [14] [15] [16] [17] Островки CpG, обнаруженные в областях промотора, обычно защищены от метилирования ДНК. В раковых клетках CpG-островки гипометилированы. [20] В регионах, окружающих острова CpG, называемых берегами островов CpG, происходит большая часть метилирования ДНК в контексте динуклеотидов CpG. Раковые клетки почтительно метилируются на берегах островов CpG. В раковых клетках гиперметилирование на берегах островков CpG перемещается в островки CpG, или гипометилирование островков CpG перемещается на берега островков CpG, устраняя резкие эпигенетические границы между этими генетическими элементами. [21] В раковых клетках «глобальное гипометилирование» из-за нарушения ДНК-метилтрансфераз (DNMT) может способствовать митотической рекомбинации и перестройке хромосом , что в конечном итоге приводит к анеуплоидии , когда хромосомы не могут правильно разделиться во время митоза . [14] [15] [16] [17]
Метилирование CpG-островков важно для регуляции экспрессии генов, однако метилирование цитозина может непосредственно приводить к дестабилизирующим генетическим мутациям и предраковому состоянию клеток. Метилированные цитозины делают гидролиз аминогруппы . и спонтанное превращение в тимин более благоприятным Они могут вызывать аберрантное рекрутирование белков хроматина . Метилирование цитозина изменяет степень поглощения ультрафиолетового света нуклеотидным основанием, создавая пиримидиновые димеры . Когда мутация приводит к потере гетерозиготности в сайтах генов-супрессоров опухоли, эти гены могут стать неактивными. Мутации одной пары оснований во время репликации также могут иметь пагубные последствия. [12]
Модификация гистонов
[ редактировать ]Эукариотическая ДНК имеет сложную структуру. Обычно он обернут вокруг специальных белков, называемых гистонами, образуя структуру, называемую нуклеосомой . Нуклеосома состоит из 2 наборов по 4 гистона: H2A , H2B , H3 и H4 . Кроме того, гистон H1 способствует упаковке ДНК вне нуклеосомы. Определенные ферменты, модифицирующие гистоны, могут добавлять или удалять функциональные группы к гистонам, и эти модификации влияют на уровень транскрипции генов, обернутых вокруг этих гистонов, и уровень репликации ДНК. Профили модификации гистонов здоровых и раковых клеток имеют тенденцию различаться.
По сравнению со здоровыми клетками, в раковых клетках наблюдается пониженное содержание моноацетилированных и триметилированных форм гистона H4 (снижение H4ac и H4me3). [22] Кроме того, мышиные модели показали, что снижение асимметричного диметилирования гистона H4R3 (H4R3me2a) промотора p19ARF коррелирует с более поздними случаями онкогенеза и метастазирования. [23] В мышиных моделях потеря ацетилирования и триметилирования гистона H4 увеличивается по мере продолжения роста опухоли. [22] Потеря ацетилирования гистона H4 лизина 16 ( H4K16ac ), которая является признаком старения теломер , приводит к потере ацетилирования. Некоторые ученые надеются, что с этой конкретной потерей ацетилирования гистонов можно бороться с помощью ингибитора гистондеацетилазы (HDAC), специфичного для SIRT1 , HDAC, специфичного для H4K16. [10] [24]
Другие гистоновые метки, связанные с онкогенезом, включают повышенное деацетилирование (снижение ацетилирования) гистонов H3 и H4, снижение триметилирования гистона H3 лизина 4 ( H3K4me3 ), а также повышенное монометилирование гистона H3 лизина 9 ( H3K9me1 ) и триметилирование гистона H3 лизина 27 ( H3K27me3). ). Эти модификации гистонов могут заставить замолчать гены-супрессоры опухолей, несмотря на снижение метилирования CpG-островка гена (событие, которое обычно активирует гены). [25] [26]
Некоторые исследования были сосредоточены на блокировании действия BRD4 на ацетилированные гистоны, что, как было показано, увеличивает экспрессию белка Myc , участвующего в некоторых видах рака. Процесс разработки препарата, связывающегося с BRD4, примечателен совместным и открытым подходом, который использует команда. [27]
Ген-супрессор опухоли p53 регулирует репарацию ДНК и может вызывать апоптоз в клетках с нарушением регуляции. Э. Сото-Рейес и Ф. Рециллас-Тарга выяснили важность белка CTCF в регуляции экспрессии р53. [28] CTCF, или фактор связывания CCCTC, представляет собой белок «цинковых пальцев» , который изолирует промотор р53 от накопления репрессивных гистоновых меток. В некоторых типах раковых клеток белок CTCF не связывается нормально, а промотор р53 накапливает репрессивные гистоновые метки, вызывая снижение экспрессии р53. [28]
Также могут возникать мутации в самом эпигенетическом механизме, потенциально ответственные за изменение эпигенетических профилей раковых клеток. Варианты гистонов семейства H2A высококонсервативны у млекопитающих и играют решающую роль в регуляции многих ядерных процессов путем изменения структуры хроматина . Один из ключевых вариантов H2A, H2A.X, отмечает повреждение ДНК, способствуя привлечению белков репарации ДНК для восстановления целостности генома. Другой вариант, H2A.Z, играет важную роль как в активации, так и в репрессии генов. Высокий уровень экспрессии H2A.Z обнаруживается при многих видах рака и в значительной степени связан с клеточной пролиферацией и геномной нестабильностью. [11] Вариант гистона макроH2A1 важен в патогенезе многих типов рака, например, гепатоцеллюлярной карциномы. [29] Другие механизмы включают снижение H4K16ac, которое может быть вызвано либо снижением активности ацетилтрансфераз гистонов (HAT), либо увеличением деацетилирования с помощью SIRT1. [10] Аналогичным образом, инактивирующая мутация сдвига рамки считывания в HDAC2 , деацетилазе гистонов хвоста гистонов , которая действует на многие лизины , была связана с раком, демонстрирующим измененные паттерны ацетилирования гистонов. [30] Эти результаты указывают на многообещающий механизм изменения эпигенетических профилей посредством ферментативного ингибирования или усиления. Новой развивающейся областью, которая фиксирует токсикологические эпигенетические изменения в результате воздействия различных соединений (лекарств, продуктов питания и окружающей среды), является токсикоэпигенетика. В этой области растет интерес к картированию изменений модификаций гистонов и их возможных последствий. [31]
Повреждение ДНК , вызванное ультрафиолетовым излучением, ионизирующим излучением , токсинами окружающей среды и метаболическими химическими веществами, также может привести к геномной нестабильности и раку. Реакция повреждения ДНК на двухцепочечные разрывы ДНК (DSB) частично опосредована модификациями гистонов. В DSB белковый комплекс MRE11 - RAD50 - NBS1 (MRN) рекрутирует киназу с мутацией атаксии телеангиэктазии (ATM), которая фосфорилирует серин 129 гистона 2A. MDC1, медиатор контрольной точки 1 повреждения ДНК, связывается с фосфопептидом, и фосфорилирование H2AX может распространяться за счет петли положительной обратной связи рекрутирования и фосфорилирования MRN-ATM. TIP60 ацетилирует γH2AX , который затем полиубиквитилируется . RAP80 , субъединица белкового комплекса чувствительности к раку молочной железы 1-го типа, восстанавливающего ДНК ( BRCA1 -A), связывает убиквитин, прикрепленный к гистонам. Активность BRCA1-A останавливает клеточный цикл в контрольной точке G2/M , давая время для восстановления ДНК, иначе может быть инициирован апоптоз . [32]
Замалчивание генов микроРНК
[ редактировать ]У млекопитающих микроРНК (миРНК) регулируют около 60% транскрипционной активности генов, кодирующих белки. [33] Некоторые микроРНК также подвергаются связанному с метилированием молчанию в раковых клетках. [34] [35] Let-7 и miR15/16 играют важную роль в подавлении RAS и BCL2 онкогенов , а их подавление происходит в раковых клетках. [18] Снижение экспрессии миР-125b1, миРНК, которая действует как супрессор опухоли , наблюдалось при раке простаты, яичников , молочной железы и глиальных клеток. Эксперименты in vitro показали, что миР-125b1 нацелена на два гена, HER2/neu и ESR1 , которые связаны с раком молочной железы. Метилирование ДНК, в частности гиперметилирование, является одним из основных способов эпигенетического подавления миР-125b1. У больных раком молочной железы наблюдалось гиперметилирование CpG-островков, расположенных проксимальнее места начала транскрипции. Потеря связывания CTCF и увеличение репрессивных меток гистонов, H3K9me3 и H3K27me3, коррелируют с метилированием ДНК и молчанием miR-125b1. Механистически CTCF может функционировать как пограничный элемент, останавливающий распространение метилирования ДНК. Результаты экспериментов, проведенных Soto-Reyes et al. [36] указывают на негативное влияние метилирования на функцию и экспрессию миР-125b1. Таким образом, они пришли к выводу, что метилирование ДНК играет роль в подавлении гена. Более того, некоторые микроРНК эпигенетически подавляются на ранних стадиях рака молочной железы, и поэтому эти микроРНК потенциально могут быть полезны в качестве опухолевых маркеров. [36] Эпигенетическое молчание генов микроРНК вследствие аберрантного метилирования ДНК является частым явлением в раковых клетках; почти одна треть промоторов микроРНК, активных в нормальных клетках молочной железы, была обнаружена гиперметилированной в клетках рака молочной железы – это в несколько раз больше, чем обычно наблюдается для генов, кодирующих белки. [37]
Метаболическое перекодирование эпигенетики при раке
[ редактировать ]Нарушение регуляции метаболизма позволяет опухолевым клеткам генерировать необходимые строительные блоки, а также модулировать эпигенетические метки, поддерживающие возникновение и прогрессирование рака. Метаболические изменения, вызванные раком, изменяют эпигенетический ландшафт, особенно модификации гистонов и ДНК, тем самым способствуя злокачественной трансформации, адаптации к недостаточному питанию и метастазированию. Чтобы удовлетворить биосинтетические потребности раковых клеток, метаболические пути изменяются путем одновременного манипулирования онкогенами и генами, подавляющими опухоль. [38] Накопление определенных метаболитов при раке может воздействовать на эпигенетические ферменты, чтобы глобально изменить эпигенетический ландшафт. Метаболические изменения, связанные с раком, приводят к локус-специфическому перекодированию эпигенетических меток. Эпигенетика рака может быть точно перепрограммирована клеточным метаболизмом посредством 1) дозозависимой модуляции эпигенетики рака метаболитами; 2) специфическое рекрутирование метаболических ферментов; и 3) нацеливание на эпигенетические ферменты с помощью сигналов питания. [38] Помимо модуляции метаболического программирования на молекулярном уровне, существуют факторы микроокружения, которые могут влиять на метаболическое перекодирование. Эти влияния включают питательный, воспалительный и иммунный ответ злокачественных тканей.
Репарация микроРНК и ДНК
[ редактировать ]Повреждение ДНК, по-видимому, является основной причиной рака. [39] [40] Если репарация ДНК недостаточна, повреждения ДНК имеют тенденцию накапливаться. Такое избыточное повреждение ДНК может увеличить мутационные ошибки во время репликации ДНК из-за склонного к ошибкам синтеза транслезий . Чрезмерное повреждение ДНК также может усилить эпигенетические изменения из-за ошибок во время восстановления ДНК. [41] [42] Такие мутации и эпигенетические изменения могут привести к раку (см. злокачественные новообразования ).
Зародышевые мутации в генах репарации ДНК вызывают только 2–5% случаев рака толстой кишки . [43] Однако измененная экспрессия микроРНК, вызывающая нарушения репарации ДНК, часто связана с раком и может быть важным причинным фактором этих видов рака.
Избыточная экспрессия некоторых микроРНК может напрямую снижать экспрессию специфических белков репарации ДНК. Ван и др. [44] относятся к 6 генам репарации ДНК, на которые непосредственно нацелены микроРНК, указанные в скобках: ATM (миР-421), RAD52 (миР-210, миР-373), RAD23B (миР-373), MSH2 (миР-21), BRCA1 (миР-182) и P53 (миР-504, миР-125b). Совсем недавно Тесситоре и др. [45] перечислены дополнительные гены репарации ДНК, на которые непосредственно нацелены дополнительные микроРНК, включая ATM (миР-18а, миР-101), DNA-PK (миР-101), ATR (миР-185), Wip1 (миР-16), MLH1, MSH2 и MSH6 (миР-155), ERCC3 и ERCC4 (миР-192) и UNG2 (миР-16, миР-34c и миР-199a). Среди этих микроРНК миР-16, миР-18а, миР-21, миР-34c, миР-125b, миР-101, миР-155, миР-182, миР-185 и миР-192 относятся к числу идентифицированных Шнекенбургером и Дидерих [46] как сверхэкспрессированный при раке толстой кишки вследствие эпигенетического гипометилирования. Чрезмерная экспрессия любой из этих микроРНК может вызвать снижение экспрессии целевого гена репарации ДНК.
До 15% дефицитов MLH1 при спорадическом раке толстой кишки, по-видимому, обусловлено сверхэкспрессией микроРНК миР-155 , которая подавляет экспрессию MLH1. [47] Однако в большинстве из 68 спорадических случаев рака толстой кишки со сниженной экспрессией белка несоответствия ДНК репарации MLH1 был обнаружен дефицит из-за эпигенетического метилирования CpG-островка гена MLH1 . [48]
В 28% глиобластом белок репарации ДНК MGMT недостаточен, но промотор MGMT не метилирован. [49] В глиобластомах без метилированных промоторов MGMT уровень микроРНК miR-181d обратно коррелирует с экспрессией белка MGMT, а прямой мишенью miR-181d является 3'UTR мРНК MGMT ( три основных нетранслируемых участка мРНК MGMT). [49] Таким образом, в 28% глиобластом причинным фактором может быть повышенная экспрессия миР-181d и сниженная экспрессия фермента репарации ДНК MGMT. В 29–66% [49] [50] В глиобластомах репарация ДНК недостаточна из-за эпигенетического метилирования гена MGMT , что снижает экспрессию белка MGMT.
Белки группы A с высокой подвижностью ( HMGA ), характеризующиеся AT-крючком , представляют собой небольшие негистоновые, ассоциированные с хроматином белки, которые могут модулировать транскрипцию. МикроРНК контролируют экспрессию белков HMGA , и эти белки ( HMGA1 и HMGA2 ) являются элементами, контролирующими транскрипцию архитектурного хроматина. Палмиери и др. [51] показали, что в нормальных тканях гены HGMA1 и HMGA2 подвергаются воздействию (и, таким образом, сильно снижают экспрессию) миР-15 , миР-16 , миР-26а , миР-196а2 и Let-7a .
Экспрессия HMGA практически не обнаруживается в дифференцированных тканях взрослых, но повышена при многих видах рака. Белки HGMA представляют собой полипептиды из ~100 аминокислотных остатков, характеризующиеся модульной организацией последовательностей. Эти белки имеют три сильно положительно заряженных участка, называемых АТ-крючками , которые связывают малую бороздку богатых АТ участков ДНК в определенных участках ДНК. Неоплазии человека, включая рак щитовидной железы, предстательной железы, шейки матки, колоректальный рак, рак поджелудочной железы и яичников, демонстрируют значительное увеличение белков HMGA1a и HMGA1b. [52] У трансгенных мышей с HMGA1, нацеленным на лимфоидные клетки, развивается агрессивная лимфома, показывая, что высокая экспрессия HMGA1 не только связана с раком, но и что ген HMGA1 может действовать как онкоген, вызывая рак. [53] Бальдасарре и др., [54] показали, что белок HMGA1 связывается с промоторной областью гена репарации ДНК BRCA1 и ингибирует активность промотора BRCA1 . Они также показали, что, хотя только 11% опухолей молочной железы имели гиперметилирование гена BRCA1 , 82% агрессивных видов рака молочной железы имеют низкую экспрессию белка BRCA1, и большинство этих снижений было связано с ремоделированием хроматина за счет высоких уровней белка HMGA1.
Белок HMGA2 специфически нацелен на промотор ERCC1 , тем самым снижая экспрессию этого гена репарации ДНК. [55] Экспрессия белка ERCC1 была недостаточной в 100% из 47 оцененных случаев рака толстой кишки (хотя степень участия HGMA2 неизвестна). [56]
Палмиери и др. [51] показали, что каждая из микроРНК, нацеленных на гены HMGA , резко снижается почти во всех изученных аденомах гипофиза человека по сравнению с нормальным гипофизом. В соответствии со снижением регуляции этих микроРНК, нацеленных на HMGA, наблюдалось увеличение количества специфичных для HMGA1 и HMGA2 мРНК. Три из этих микроРНК (миР-16, миР-196а и Let-7a) [46] [57] имеют метилированные промоторы и, следовательно, низкую экспрессию при раке толстой кишки. Для двух из них, миР-15 и миР-16, кодирующие области эпигенетически молчат при раке из-за активности гистондеацетилазы . [58] Когда эти микроРНК экспрессируются на низком уровне, белки HMGA1 и HMGA2 экспрессируются на высоком уровне. (снижают экспрессию) HMGA1 и HMGA2 нацелены на гены репарации ДНК BRCA1 и ERCC1 . Таким образом, репарация ДНК может быть снижена, что, вероятно, способствует прогрессированию рака. [40]
Пути репарации ДНК
[ редактировать ]
В таблице в этом разделе показаны некоторые часто встречающиеся агенты, повреждающие ДНК, примеры повреждений ДНК, которые они вызывают, и пути, которые устраняют эти повреждения ДНК. По крайней мере 169 ферментов либо непосредственно участвуют в репарации ДНК, либо влияют на процессы репарации ДНК. [59] Из них 83 непосредственно задействованы в восстановлении 5 типов повреждений ДНК, показанных на диаграмме.
Некоторые из наиболее хорошо изученных генов, играющих центральную роль в процессах восстановления, показаны на диаграмме. Обозначения генов, выделенные красным, серым или голубым цветом, указывают на гены, которые часто эпигенетически изменяются при различных типах рака. Статьи в Википедии о каждом гене, выделенном красным, серым или голубым, описывают эпигенетические изменения и рак, при которых обнаруживаются эти эпимутации. Две обширные экспериментальные обзорные статьи [60] [61] также документируют большинство этих нарушений репарации эпигенетической ДНК при раке.
Гены, выделенные красным, часто редуцируются или подавляются эпигенетическими механизмами при различных видах рака. Когда экспрессия этих генов низкая или отсутствует, повреждения ДНК могут накапливаться. Ошибки репликации, выходящие за рамки этих повреждений (см. Синтез транслезий ), могут привести к увеличению количества мутаций и, в конечном итоге, к раку. Эпигенетическая репрессия генов репарации ДНК в точных путях репарации ДНК, по-видимому, играет центральную роль в канцерогенезе .
Два выделенных серым гена RAD51 и BRCA2 необходимы для гомологичной рекомбинационной репарации. Иногда они эпигенетически сверхэкспрессируются, а иногда и недостаточно экспрессируются при некоторых видах рака. Как указано в статьях Википедии о RAD51 и BRCA2 , такие виды рака обычно имеют эпигенетический дефицит других генов репарации ДНК. Эти недостатки репарации, вероятно, приведут к увеличению количества невосстановленных повреждений ДНК. Сверхэкспрессия RAD51 и BRCA2, наблюдаемая при этих видах рака, может отражать селективное давление на компенсаторную RAD51 или BRCA2 сверхэкспрессию и усиление гомологичной рекомбинационной репарации, чтобы, по крайней мере, частично справиться с такими избыточными повреждениями ДНК. В тех случаях, когда RAD51 или BRCA2 недостаточно экспрессируются, это само по себе может привести к увеличению невосстановленных повреждений ДНК. Ошибки репликации, выходящие за рамки этих повреждений (см. «Синтез транслезий »), могут вызвать усиление мутаций и рак, так что недостаточная экспрессия RAD51 или BRCA2 сама по себе будет канцерогенной.
Гены, выделенные голубым цветом, участвуют в пути соединения концов, опосредованном микрогомологией (MMEJ), и их активация повышается при раке. MMEJ — это дополнительный, подверженный ошибкам и неточный путь восстановления двухцепочечных разрывов. При репарации двухцепочечного разрыва MMEJ гомология 5-25 комплементарных пар оснований между обеими парными цепями достаточна для выравнивания цепей, но обычно присутствуют несовпадающие концы (лоскуты). MMEJ удаляет лишние нуклеотиды (лоскуты) в местах соединения цепей, а затем лигирует нити, чтобы создать неповрежденную двойную спираль ДНК. MMEJ почти всегда включает хотя бы небольшую делецию, так что это мутагенный путь. [62] FEN1 , эндонуклеаза лоскута в MMEJ, эпигенетически увеличивается за счет гипометилирования промотора и сверхэкспрессируется в большинстве случаев рака молочной железы. [63] простата, [64] желудок, [65] [66] нейробластомы, [67] поджелудочная железа, [68] и легкое. [69] PARP1 также сверхэкспрессируется, когда сайт ETS его промоторной области эпигенетически гипометилирован, и это способствует прогрессированию рака эндометрия. [70] рак яичников с мутацией BRCA, [71] и серозный рак яичников с мутацией BRCA. [72] Другие гены пути MMEJ также сверхэкспрессируются при ряде видов рака (краткую информацию см. в MMEJ ) и также показаны синим цветом.
Частоты эпимутаций в генах репарации ДНК
[ редактировать ]Дефицит белков репарации ДНК, которые функционируют в точных путях репарации ДНК, увеличивает риск мутаций. Частота мутаций сильно увеличивается в клетках с мутациями в репарации несоответствия ДНК. [73] [74] или в гомологичной рекомбинационной репарации (HRR). [75] Лица с унаследованными мутациями в любом из 34 генов репарации ДНК подвергаются повышенному риску развития рака (см. Дефекты репарации ДНК и повышенный риск рака ).
При спорадическом раке иногда обнаруживается, что дефицит репарации ДНК обусловлен мутацией гена репарации ДНК, но гораздо чаще снижение или отсутствие экспрессии генов репарации ДНК происходит из-за эпигенетических изменений, которые уменьшают или подавляют экспрессию генов. Например, из 113 последовательно исследованных случаев колоректального рака только четыре имели миссенс-мутацию в гене репарации ДНК MGMT , в то время как у большинства наблюдалось снижение экспрессии MGMT из-за метилирования области промотора MGMT (эпигенетическое изменение). [76] Аналогичным образом, из 119 случаев колоректального рака с дефицитом репарации ошибочного спаривания, при котором отсутствовала экспрессия гена репарации ДНК PMS2 , белок PMS2 был дефицитным в 6 из-за мутаций в гене PMS2 , тогда как в 103 случаях экспрессия PMS2 была недостаточной, поскольку его партнер по спариванию MLH1 был подавлен. из-за метилирования промотора (белок PMS2 нестабильен в отсутствие MLH1). [48] В других 10 случаях потеря экспрессии PMS2, вероятно, была связана с эпигенетической сверхэкспрессией микроРНК, миР-155, которая подавляет MLH1. [47]
Эпигенетические дефекты в генах репарации ДНК часто встречаются при раке. В таблице множественные виды рака оценивались на предмет снижения или отсутствия экспрессии интересующего гена репарации ДНК, а показанная частота представляет собой частоту, с которой рак имел эпигенетический дефицит экспрессии генов. Такие эпигенетические дефекты, вероятно, возникают на ранних стадиях канцерогенеза , поскольку они также часто обнаруживаются (хотя и с несколько меньшей частотой) в дефекте поля , окружающем рак, из которого рак, вероятно, возник (см. Таблицу).
| Рак | Ген | Частота в Раке | Частота в дефекте поля | Ссылка. |
|---|---|---|---|---|
| Колоректальный | МГМТ | 46% | 34% | [77] |
| Колоректальный | МГМТ | 47% | 11% | [78] |
| Колоректальный | МГМТ | 70% | 60% | [79] |
| Колоректальный | МШ2 | 13% | 5% | [78] |
| Колоректальный | ЭРСС1 | 100% | 40% | [56] |
| Колоректальный | ПМС2 | 88% | 50% | [56] |
| Колоректальный | XPF | 55% | 40% | [56] |
| Голова и шея | МГМТ | 54% | 38% | [80] |
| Голова и шея | МЛХ1 | 33% | 25% | [81] |
| Голова и шея | МЛХ1 | 31% | 20% | [82] |
| Желудок | МГМТ | 88% | 78% | [83] |
| Желудок | МЛХ1 | 73% | 20% | [84] |
| Желудок | ЭРСС1 | 95-100% | 14-65% | [85] |
| Желудок | ПМС2 | 95-100% | 14-60% | [85] |
| пищевод | МЛХ1 | 77%–100% | 23%–79% | [86] |
Похоже, что рак часто может быть инициирован эпигенетическим снижением экспрессии одного или нескольких ферментов репарации ДНК. Снижение репарации ДНК, вероятно, приводит к накоплению повреждений ДНК. Склонный к ошибкам синтез транслейкоза мимо некоторых из этих повреждений ДНК может привести к мутации с селективным преимуществом. Клональный участок с селективным преимуществом может расти и вытеснять соседние клетки, образуя дефект поля . Хотя у клетки нет очевидного селективного преимущества в снижении репарации ДНК, эпимутация гена репарации ДНК может выполняться в качестве пассажира, когда клетки с селективно выгодной мутацией реплицируются. В клетках, несущих как эпимутацию гена репарации ДНК, так и мутацию с селективным преимуществом, будут накапливаться дальнейшие повреждения ДНК, которые, в свою очередь, могут привести к дальнейшим мутациям с еще большими селективными преимуществами. Таким образом, эпигенетические дефекты репарации ДНК могут способствовать характерной высокой частоте мутаций в геномах раковых заболеваний и вызывать их канцерогенное прогрессирование.
Рак имеет высокий уровень нестабильности генома , связанный с высокой частотой мутаций . Высокая частота геномных мутаций увеличивает вероятность возникновения определенных мутаций, которые активируют онкогены и инактивируют гены-супрессоры опухолей, приводя к канцерогенезу . На основе полногеномного секвенирования обнаружено, что раковые клетки имеют от тысяч до сотен тысяч мутаций во всем геноме. [87] (Также см. «Частоты мутаций при раке »). Для сравнения: частота мутаций во всем геноме между поколениями людей (от родителей к детям) составляет около 70 новых мутаций на поколение. [88] [89] В участках генома, кодирующих белок, существует всего около 0,35 мутаций между поколениями родителей и детей (менее одного мутировавшего белка на поколение). [90] Полногеномное секвенирование клеток крови пары идентичных близнецов-100-летних долгожителей выявило только 8 соматических различий, хотя соматические вариации, возникающие менее чем в 20% клеток крови, остались бы незамеченными. [91]
Хотя повреждения ДНК могут привести к мутациям из-за склонного к ошибкам синтеза транслейкоза , повреждения ДНК также могут привести к эпигенетическим изменениям во время ошибочных процессов репарации ДНК. [41] [42] [92] [93] Повреждения ДНК, которые накапливаются из-за дефектов эпигенетической репарации ДНК, могут быть источником повышенных эпигенетических изменений, обнаруженных во многих генах при раке. В раннем исследовании, изучавшем ограниченный набор транскрипционных промоторов, Fernandez et al. [94] исследовали профили метилирования ДНК 855 первичных опухолей. При сравнении каждого типа опухоли с соответствующей нормальной тканью 729 сайтов CpG-островков (55% из 1322 оцененных сайтов CpG) показали дифференциальное метилирование ДНК. Из этих сайтов 496 были гиперметилированы (репрессированы) и 233 гипометилированы (активированы). Таким образом, в опухолях наблюдается высокий уровень изменений метилирования эпигенетических промоторов. Некоторые из этих эпигенетических изменений могут способствовать прогрессированию рака.
Эпигенетические канцерогены
[ редактировать ]относят разнообразные соединения К эпигенетическим канцерогенам — они приводят к увеличению заболеваемости опухолями, но не проявляют мутагенной активности (следует также исключить токсичные соединения или патогены, вызывающие опухоли, склонные к усиленной регенерации). Примеры включают диэтилстильбэстрол , арсенит , гексахлорбензол и никеля соединения .
Многие тератогены оказывают специфическое воздействие на плод посредством эпигенетических механизмов. [95] [96] Хотя эпигенетические эффекты могут сохранять действие тератогена, такого как диэтилстильбестрол , на протяжении всей жизни больного ребенка, возможность врожденных дефектов, возникающих в результате воздействия отцов или во втором и последующих поколениях потомства, обычно отвергается на теоретических основаниях и из-за отсутствия доказательство. [97] Тем не менее, был продемонстрирован ряд аномалий, опосредованных мужчинами, и, вероятно, их будет больше. [98] В информации на этикетке FDA для видазы, препарата 5-азацитидина (неметилируемого аналога цитидина, который вызывает гипометилирование при включении в ДНК), говорится, что «мужчинам следует рекомендовать не заводить ребенка» во время использования препарата, ссылаясь на данные, полученные на обработанных самцах мышей. снижение фертильности, повышенная потеря эмбрионов и аномальное развитие эмбрионов. [99] У крыс эндокринные различия наблюдались у потомков самцов, подвергшихся воздействию морфина. [100] У мышей были описаны эффекты диэтилстильбестерола второго поколения, возникающие по эпигенетическим механизмам. [101]
Подтипы рака
[ редактировать ]Рак кожи
[ редактировать ]Меланома – это смертельный рак кожи, который развивается из меланоцитов. Известно, что несколько эпигенетических изменений играют роль в переходе меланоцитов в клетки меланомы. Сюда входит метилирование ДНК, которое может передаваться по наследству без внесения изменений в последовательность ДНК, а также подавление генов-супрессоров опухолей в эпидермисе, которые подвергались воздействию УФ-излучения в течение определенного периода времени. [102] Замалчивание генов-супрессоров опухоли приводит к фотоканцерогенезу , который связан с эпигенетическими изменениями в метилировании ДНК, ДНК-метилтрансферазах и ацетилировании гистонов. [102] Эти изменения являются следствием нарушения регуляции соответствующих ферментов. К числу этих ферментов относятся несколько гистоновых метилтрансфераз и деметилаз. [103]
Рак простаты
[ редактировать ]Ежегодно рак простаты убивает около 35 000 мужчин, и только в Северной Америке рак простаты диагностируется примерно у 220 000 мужчин в год. [104] Рак простаты является второй по значимости причиной смертности от рака среди мужчин, и в течение жизни мужчины этим заболеванием заболевает каждый шестой мужчина. [104] Изменения в ацетилировании гистонов и метилировании ДНК происходят в различных генах, влияющих на рак простаты, и наблюдались в генах, участвующих в гормональной реакции. [105] Более чем в 90% случаев рака простаты наблюдается генов за счет гиперметилирования CpG-островков промотора GSTP1 гена подавление , который защищает клетки простаты от геномного повреждения, вызванного различными окислителями или канцерогенами . [106] , специфичная для метилирования в реальном времени Полимеразная цепная реакция (ПЦР), предполагает, что многие другие гены также гиперметилированы. [106] Экспрессия генов в простате может модулироваться изменениями в питании и образе жизни. [107]
Рак шейки матки
[ редактировать ]Второй наиболее распространенной злокачественной опухолью у женщин является инвазивный рак шейки матки (ИКШ), и более 50% всех случаев инвазивного рака шейки матки (ИКШ) вызвано онконгенным вирусом папилломы человека 16 ( ВПЧ16 ). [108] Кроме того, интраэпителиальная неоплазия шейки матки (ЦИН) в первую очередь вызывается онкогенным ВПЧ16. [108] Как и во многих случаях, причинный фактор рака не всегда идет прямым путем от инфекции к развитию рака. Паттерны геномного метилирования связаны с инвазивным раком шейки матки. В регионе HPV16L1 14 протестированных сайтов CpG имеют значительно более высокое метилирование в CIN3+, чем в геномах HPV16 женщин без CIN3 . [108] Было обнаружено, что только 2 из 16 сайтов CpG, протестированных в регуляторной области выше HPV16, связаны с повышенным метилированием в CIN3+. [108] Это говорит о том, что прямой путь от инфекции к раку иногда ведет к предраковому состоянию при интраэпителиальной неоплазии шейки матки. Кроме того, повышенное метилирование сайта CpG было обнаружено на низких уровнях в большинстве из пяти изученных ядерных генов хозяина, включая 5/5 TERT , 1/4 DAPK1 , 2/5 RARB , MAL и CADM1 . [108] Более того, 1/3 сайтов CpG в митохондриальной ДНК были связаны с повышенным метилированием CIN3+. [108] Таким образом, существует корреляция между CIN3+ и повышенным метилированием сайтов CpG в открытой рамке считывания L1 HPV16. [108] Это может стать потенциальным биомаркером для будущих скринингов раковых и предраковых заболеваний шейки матки. [108]
Лейкемия
[ редактировать ]Недавние исследования показали, что ген лейкемии смешанного происхождения (MLL) вызывает лейкемию путем перестройки и слияния с другими генами в разных хромосомах, что является процессом под эпигенетическим контролем. [109] Мутации в MLL блокируют правильные регуляторные области в транслокациях или инсерциях, связанных с лейкемией, вызывая злокачественную трансформацию, контролируемую генами НОХ. [110] Именно это приводит к увеличению количества лейкоцитов. Гены, связанные с лейкемией, управляются теми же путями, которые контролируют эпигенетику, сигнальную трансдукцию, регуляцию транскрипции и энергетический метаболизм. Было указано, что причиной лейкемии могут быть инфекции, электромагнитные поля и повышенная масса тела при рождении. [111]
Саркома
[ редактировать ]Ежегодно в США регистрируется около 15 000 новых случаев саркомы, и согласно прогнозам, в 2014 году от саркомы в США умрут около 6200 человек. [112] Саркомы включают большое количество редких, гистогенетически гетерогенных мезенхимальных опухолей, которые, например, включают хондросаркому, саркому Юинга, лейомиосаркому, липосаркому, остеосаркому, синовиальную саркому и (альвеолярную и эмбриональную) рабдомиосаркому. Некоторые онкогены и гены-супрессоры опухолей эпигенетически изменяются при саркомах. К ним относятся APC, CDKN1A, CDKN2A, CDKN2B, Ezrin, FGFR1, GADD45A, MGMT, STK3, STK4, PTEN, RASSF1A, WIF1, а также несколько микроРНК. [113] Экспрессия эпигенетических модификаторов, таких как компонент BMI1 комплекса PRC1, дерегулируется при хондросаркоме, саркоме Юинга и остеосаркоме, а экспрессия компонента EZH2 комплекса PRC2 изменяется при саркоме Юинга и рабдомиосаркоме. Аналогично, экспрессия другого эпигенетического модификатора, деметилазы гистонов LSD1, увеличивается при хондросаркоме, саркоме Юинга, остеосаркоме и рабдомиосаркоме. Нацеливание лекарств и ингибирование EZH2 при саркоме Юинга, [114] или ЛСД1 при некоторых саркомах, [115] ингибирует рост опухолевых клеток в этих саркомах.
Рак легких
[ редактировать ]Рак легких является вторым по распространенности типом рака и основной причиной смертности мужчин и женщин в Соединенных Штатах. По оценкам, от рака легких зарегистрировано около 216 000 новых случаев и 160 000 смертей. [116]
Инициирование и прогрессирование карциномы легких является результатом взаимодействия генетических, эпигенетических и экологических факторов. Большинство случаев рака легких вызваны генетическими мутациями EGFR , KRAS , STK11 (также известного как LKB1 ), TP53 (также известного как p53 ) и CDKN2A (также известного как p16 или INK4a ). [117] [118] [119] при этом наиболее распространенным типом рака легких является инактивация р16. p16 представляет собой белок-супрессор опухолей, который встречается в основном у людей. Функциональное значение мутаций было проверено на многих других видах, включая мышей, кошек, собак, обезьян и коров. Идентификация этих множественных неперекрывающихся клонов не была совершенно неожиданной, поскольку гибридизация со сниженной строгостью Зооблоттинг с тем же зондом также выявил 10-15 положительных фрагментов EcoRI у всех протестированных видов. [120]
Методы идентификации
[ редактировать ]Раньше эпигенетические профили ограничивались отдельными генами, находящимися под пристальным вниманием конкретной исследовательской группы. Однако в последнее время ученые перешли к более геномному подходу для определения полного геномного профиля раковых и здоровых клеток. [10]
Популярные подходы к измерению метилирования CpG в клетках включают:
- Бисульфитное секвенирование
- Комбинированный бисульфитный рестрикционный анализ (COBRA)
- ПЦР, специфичная для метилирования
- МетиЛайт
- Пиросеквенирование
- Геномное сканирование рестрикционных ориентиров
- ПЦР с произвольным праймированием
- Анализ HELP (обогащение крошечными фрагментами HpaII с помощью ПЦР, опосредованной лигированием)
- Иммунопреципитация хроматина ChIP-Chip с использованием антител, специфичных к белкам связывающего домена метил-CpG
- ДНК Иммунопреципитация метилированной
- Профили экспрессии генов с помощью микроматрицы ДНК : сравнение уровней мРНК из линий раковых клеток до и после лечения деметилирующим агентом
Поскольку бисульфитное секвенирование считается золотым стандартом измерения метилирования CpG, при использовании одного из других методов результаты обычно подтверждаются с помощью бисульфитного секвенирования [1].Популярные подходы к определению профилей модификации гистонов в раковых и здоровых клетках включают: [10]
- Масс-спектрометрия
- Анализ иммунопреципитации хроматина
Диагностика и прогноз
[ редактировать ]Исследователи надеются идентифицировать конкретные эпигенетические профили различных типов и подтипов рака с целью использовать эти профили в качестве инструментов для более точной и точной диагностики людей. [10] Поскольку эпигенетические профили меняются, ученые хотели бы использовать различные эпигеномные профили для определения стадии развития или уровня агрессивности конкретного рака у пациентов. Например, гиперметилирование генов, кодирующих протеинкиназу, ассоциированную со смертью (DAPK), p16 и белок эпителиальной мембраны 3 (EMP3), связано с более агрессивными формами рака легких , колоректального рака и рака головного мозга . [17] Этот тип знаний может повлиять на то, как врачи будут ставить диагнозы и выбирать лечение своих пациентов.
Еще одним фактором, который повлияет на лечение пациентов, является знание того, насколько хорошо они будут реагировать на определенные виды лечения. Персонализированные эпигеномные профили раковых клеток могут дать понимание этой области. Например, MGMT — это фермент, который обращает присоединение алкильных групп к нуклеотиду гуанину . [121] Однако алкилирование гуанина — это механизм, с помощью которого некоторые химиотерапевтические препараты действуют, разрушая ДНК и вызывая гибель клеток . [122] [123] [124] [125] Следовательно, если ген, кодирующий MGMT в раковых клетках, гиперметилирован и фактически подавлен или подавлен, химиотерапевтические препараты, действующие за счет метилирования гуанина, будут более эффективными, чем в раковых клетках, которые имеют функциональный фермент MGMT.
Эпигенетические биомаркеры также могут использоваться в качестве инструментов молекулярного прогноза. В биоптатах первичной опухоли и лимфатических узлов медиастинальных гиперметилирование CDKN2A и CDH13 служит маркером повышенного риска более быстрого рецидива рака и более высокой смертности пациентов. [126]
Уход
[ редактировать ]
Эпигенетический контроль протоонко-областей и последовательностей опухолевых супрессоров посредством конформационных изменений гистонов играет роль в формировании и прогрессировании рака. [127] Фармацевтические препараты, обращающие вспять эпигенетические изменения, могут играть роль в развитии различных видов рака. [105] [127] [128]
Недавно стало очевидно, что связь между конкретными гистотипами рака и эпигенетическими изменениями может способствовать разработке новых эпилекарств. [129] Разработка лекарств была сосредоточена главным образом на модификации ДНК-метилтрансферазы , ацетилтрансферазы гистонов (HAT) и деацетилазы гистонов (HDAC). [130]
Препараты, которые специфически воздействуют на инвертированный паттерн метилирования раковых клеток, включают ДНК-метилтрансферазы ингибиторы азацитидин. [131] [132] и децитабин . [133] [134] Эти гипометилирующие агенты используются для лечения миелодиспластического синдрома . [135] рак крови , вызванный аномальными стволовыми клетками костного мозга . [12] Эти агенты ингибируют все три типа активных ДНК-метилтрансфераз и считались высокотоксичными, но оказались эффективными при использовании в низких дозах, замедляя прогрессирование миелодиспластического синдрома в лейкемию . [136]
Ингибиторы гистондеацетилазы (HDAC) демонстрируют эффективность при лечении Т-клеточной лимфомы . два ингибитора HDAC, вориностат и ромидепсин , были одобрены Управлением по контролю за продуктами и лекарствами . [137] [138] Однако, поскольку эти ингибиторы HDAC изменяют состояние ацетилирования многих белков в дополнение к интересующему гистону, знание основного механизма на молекулярном уровне ответа пациента необходимо для повышения эффективности использования таких ингибиторов в качестве лечения. [18] Было обнаружено, что лечение ингибиторами HDAC способствует реактивации генов после того, как ингибиторы ДНК-метилтрансфераз подавляют транскрипцию. [139] Панобиностат одобрен для определенных ситуаций при миеломе . [140]
Другими фармацевтическими объектами исследований являются гистон-лизин-метилтрансферазы (KMT) и протеин-аргинин-метилтрансферазы (PRMT). [141] Доклинические исследования показали, что луназин может иметь потенциально полезные эпигенетические эффекты. [142]
Эпигенетическая терапия
[ редактировать ]Эпигенетическая терапия рака оказалась многообещающим и возможным методом лечения раковых клеток. Эпигенетическая инактивация является идеальной мишенью для раковых клеток, поскольку она нацелена на гены, необходимые для контроля роста клеток, особенно роста раковых клеток. Крайне важно, чтобы эти гены были реактивированы, чтобы подавить рост опухоли и повысить чувствительность клеток к методам лечения рака. [143] Типичная химиотерапия направлена на уничтожение и уничтожение раковых клеток в организме. Рак, вызванный генетическими изменениями клеток, обычно необратим, и его практически невозможно обратить вспять. Это отличается от эпигенетического рака, поскольку рак, вызывающий эпигенетические аберрации, имеет возможность обратить вспять и вернуть клетки к нормальному функционированию. Способность обращать вспять эпигенетические механизмы объясняется тем фактом, что кодирование генов, подавляемых посредством модификации гистонов и ДНК, не изменяется. [144]
Существует два основных типа эпигенетических изменений в раковых клетках: метилирование ДНК и модификация гистонов. Целью эпигенетической терапии является подавление этих изменений. ДНК-метилтрансферазы (DNMT) и деацетилазы гистонов (HDAC) являются основными катализаторами эпигенетических модификаций раковых клеток. [145] Цель эпигенетической терапии — подавить это метилирование и обратить вспять эти модификации, чтобы создать новый эпигеном, в котором раковые клетки больше не процветают, а подавление опухоли является новой функцией. Синтетические лекарства используются в качестве инструментов эпигенетической терапии из-за их способности ингибировать ферменты, вызывающие модификации гистонов и метилирование ДНК. Комбинированная терапия — это один из методов эпигенетической терапии, который включает использование более чем одного синтетического препарата. Эти препараты включают в себя низкие дозы ингибитора DNMT, а также ингибитор HDAC. Вместе эти препараты способны воздействовать на связь между метилированием ДНК и модификацией гистонов. [146]
Цель эпигенетической терапии рака в отношении метилирования ДНК состоит в том, чтобы уменьшить метилирование ДНК и, в свою очередь, уменьшить молчание генов, связанных с подавлением опухоли. [147] Термин, связанный со снижением метилирования ДНК, будет известен как гипометилирование. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) в настоящее время одобрило один гипометилирующий агент, который в результате проведения клинических испытаний показал многообещающие результаты при использовании для лечения пациентов с миелодиспластическим синдромом (МДС). [148] Этот гипометилирующий агент известен как потрясающий аналог 5-азацитидина и способствует гипометилированию, нацеливая на деградацию все ДНК-метилтрансферазы. [147]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Шарма С., Келли Т.К., Джонс П.А. (январь 2010 г.). «Эпигенетика рака» . Канцерогенез . 31 (1): 27–36. дои : 10.1093/carcin/bgp220 . ПМК 2802667 . ПМИД 19752007 .
- ^ Фогельштейн Б., Пападопулос Н., Велкулеску В.Е., Чжоу С., Диас Л.А., Кинцлер К.В. (март 2013 г.). «Пейзажи генома рака» . Наука . 339 (6127): 1546–1558. Бибкод : 2013Sci...339.1546V . дои : 10.1126/science.1235122 . ПМК 3749880 . ПМИД 23539594 .
- ^ Иллингворт Р.С., Грюневальд-Шнайдер У., Уэбб С., Керр А.Р., Джеймс К.Д., Тернер Д.Д. и др. (сентябрь 2010 г.). «Островки-сироты CpG идентифицируют многочисленные консервативные промоторы в геноме млекопитающих» . ПЛОС Генетика . 6 (9): e1001134. дои : 10.1371/journal.pgen.1001134 . ПМЦ 2944787 . ПМИД 20885785 .
- ^ Вэй Дж, Ли Г, Данг С, Чжоу Ю, Цзэн К, Лю М (2016). «Открытие и проверка гиперметилированных маркеров колоректального рака» . Маркеры заболеваний . 2016 : 2192853. дои : 10.1155/2016/2192853 . ПМЦ 4963574 . ПМИД 27493446 .
- ^ Беггс А.Д., Джонс А., Эль-Бахрави М., Эль-Бахвари М., Абулафи М., Ходжсон С.В., Томлинсон И.П. (апрель 2013 г.). «Полногеномный анализ метилирования доброкачественных и злокачественных колоректальных опухолей» . Журнал патологии . 229 (5): 697–704. дои : 10.1002/путь.4132 . ПМЦ 3619233 . ПМИД 23096130 .
- ^ Новак К. (декабрь 2004 г.). «Эпигенетические изменения в раковых клетках» . МедГенМед . 6 (4):17. ПМК 1480584 . ПМИД 15775844 .
- ^ Банно К., Кису И., Янокура М., Цудзи К., Масуда К., Уэки А. и др. (сентябрь 2012 г.). «Эпимутация и рак: новый канцерогенный механизм синдрома Линча (обзор)» . Международный журнал онкологии . 41 (3): 793–797. дои : 10.3892/ijo.2012.1528 . ПМЦ 3582986 . ПМИД 22735547 .
- ^ Птица А (январь 2002 г.). «Схемы метилирования ДНК и эпигенетическая память» . Гены и развитие . 16 (1): 6–21. дои : 10.1101/gad.947102 . ПМИД 11782440 .
- ^ Герман Дж.Г., Графф Дж.Р., Мёханен С., Нелькин Б.Д., Байлин С.Б. (сентябрь 1996 г.). «ПЦР, специфичная для метилирования: новый ПЦР-анализ статуса метилирования островков CpG» . Труды Национальной академии наук Соединенных Штатов Америки . 93 (18): 9821–9826. Бибкод : 1996PNAS...93.9821H . дои : 10.1073/pnas.93.18.9821 . ПМЦ 38513 . ПМИД 8790415 .
- ^ Jump up to: а б с д и ж г час Эстеллер М. (апрель 2007 г.). «Эпигеномика рака: метиломы ДНК и карты модификации гистонов». Обзоры природы. Генетика . 8 (4): 286–298. дои : 10.1038/nrg2005 . ПМИД 17339880 . S2CID 4801662 .
- ^ Jump up to: а б Вонг, Северная Каролина, Крейг Дж. М. (2011). Эпигенетика: Справочное руководство . Норфолк, Англия: Caister Academic Press. ISBN 978-1-904455-88-2 .
- ^ Jump up to: а б с Джонс, Пенсильвания, Бэйлин С.Б. (июнь 2002 г.). «Фундаментальная роль эпигенетических событий при раке». Обзоры природы. Генетика . 3 (6): 415–428. дои : 10.1038/nrg816 . ПМИД 12042769 . S2CID 2122000 .
- ^ Де Карвалью Д.Д., Шарма С., Ю Дж.С., Су С.Ф., Таберли ПК, Келли Т.К. и др. (май 2012 г.). «Скрининг метилирования ДНК идентифицирует эпигенетические события, способствующие выживанию раковых клеток» . Раковая клетка . 21 (5): 655–667. дои : 10.1016/j.ccr.2012.03.045 . ПМК 3395886 . ПМИД 22624715 .
- ^ Jump up to: а б с Герман Дж.Г. , Байлин С.Б. (ноябрь 2003 г.). «Отключение генов при раке в связи с гиперметилированием промотора». Медицинский журнал Новой Англии . 349 (21): 2042–2054. дои : 10.1056/NEJMra023075 . PMID 14627790 .
- ^ Jump up to: а б с Фейнберг А.П., Тыко Б (февраль 2004 г.). «История эпигенетики рака». Обзоры природы. Рак . 4 (2): 143–153. дои : 10.1038/nrc1279 . ПМИД 14732866 . S2CID 31655008 .
- ^ Jump up to: а б с Эггер Г., Лян Г., Апарисио А., Джонс П.А. (май 2004 г.). «Эпигенетика болезней человека и перспективы эпигенетической терапии». Природа . 429 (6990): 457–463. Бибкод : 2004Natur.429..457E . дои : 10.1038/nature02625 . ПМИД 15164071 . S2CID 4424126 .
- ^ Jump up to: а б с д Эстеллер М (2005). «Аберрантное метилирование ДНК как механизм, вызывающий рак». Ежегодный обзор фармакологии и токсикологии . 45 : 629–656. doi : 10.1146/annurev.pharmtox.45.120403.095832 . ПМИД 15822191 .
- ^ Jump up to: а б с Бэйлин С.Б., Джонс, Пенсильвания (сентябрь 2011 г.). «Десятилетие изучения эпигенома рака – биологические и трансляционные последствия» . Обзоры природы. Рак . 11 (10): 726–734. дои : 10.1038/nrc3130 . ПМК 3307543 . ПМИД 21941284 .
- ^ Эллермейер С., Хигучи Э.К., Фаднис Н., Холм Л., Джилхуд Дж.Л., Тон Г., Смит Г.Р. (май 2010 г.). «РНКи и гетерохроматин подавляют центромерную мейотическую рекомбинацию» . Труды Национальной академии наук Соединенных Штатов Америки . 107 (19): 8701–8705. Бибкод : 2010PNAS..107.8701E . дои : 10.1073/pnas.0914160107 . ПМЦ 2889303 . ПМИД 20421495 .
- ^ Эстеллер М. (апрель 2007 г.). «Эпигеномика рака: метиломы ДНК и карты модификации гистонов». Обзоры природы. Генетика . 8 (4): 286–298. дои : 10.1038/nrg2005 . ПМИД 17339880 . S2CID 4801662 .
- ^ Тимп В., Фейнберг А.П. (июль 2013 г.). «Рак как нерегулируемый эпигеном, обеспечивающий преимущество клеточного роста за счет хозяина» . Обзоры природы. Рак . 13 (7): 497–510. дои : 10.1038/nrc3486 . ПМЦ 4636434 . ПМИД 23760024 .
- ^ Jump up to: а б Фрага М.Ф., Баллестар Э., Вильяр-Гареа А., Буа-Шорне М., Эспада Дж., Шотта Г. и др. (апрель 2005 г.). «Потеря ацетилирования Lys16 и триметилирования Lys20 гистона H4 является распространенным признаком рака человека». Природная генетика . 37 (4): 391–400. дои : 10.1038/ng1531 . ПМИД 15765097 . S2CID 27245550 .
- ^ Апреликова О, Чен К., Эль Туни Л.Х., Бриньяц-Гиттард С., Хань Дж., Цю Т. и др. (апрель 2016 г.). «Эпигенетический модификатор JMJD6 амплифицируется в опухолях молочной железы и взаимодействует с c-Myc, усиливая клеточную трансформацию, прогрессирование опухоли и метастазирование» . Клиническая эпигенетика . 8 (38): 38. дои : 10.1186/s13148-016-0205-6 . ПМЦ 4831179 . ПМИД 27081402 .
- ^ Данг В., Штеффен К.К., Перри Р., Дорси Дж.А., Джонсон Ф.Б., Шилатифард А. и др. (июнь 2009 г.). «Ацетилирование гистона H4 лизина 16 регулирует продолжительность жизни клеток» . Природа . 459 (7248): 802–807. Бибкод : 2009Natur.459..802D . дои : 10.1038/nature08085 . ПМК 2702157 . ПМИД 19516333 .
- ^ Вире Э., Бреннер С., Деплюс Р., Бланшон Л., Фрага М., Дидело С. и др. (февраль 2006 г.). «Белок группы Polycomb EZH2 напрямую контролирует метилирование ДНК». Природа . 439 (7078): 871–874. Бибкод : 2006Natur.439..871V . дои : 10.1038/nature04431 . ПМИД 16357870 . S2CID 4409726 .
- ^ Ришон В.М., Сандхофф Т.В., Рифкинд Р.А., Маркс П.А. (август 2000 г.). «Ингибитор гистондеацетилазы избирательно индуцирует экспрессию p21WAF1 и ген-ассоциированное ацетилирование гистонов» . Труды Национальной академии наук Соединенных Штатов Америки . 97 (18): 10014–10019. Бибкод : 2000PNAS...9710014R . дои : 10.1073/pnas.180316197 . JSTOR 123305 . ПМК 27656 . ПМИД 10954755 .
- ^ Максмен А (август 2012 г.). «Исследование рака: открытые амбиции» . Природа . 488 (7410): 148–150. Бибкод : 2012Natur.488..148M . дои : 10.1038/488148a . ПМИД 22874946 .
- ^ Jump up to: а б Сото-Рейес Э., Ресиллас-Тарга Ф (апрель 2010 г.). «Эпигенетическая регуляция промотора гена p53 человека с помощью фактора транскрипции CTCF в трансформированных клеточных линиях». Онкоген . 29 (15): 2217–2227. дои : 10.1038/onc.2009.509 . ПМИД 20101205 . S2CID 23983571 .
- ^ Раппа Ф., Греко А., Подрини С., Каппелло Ф., Фоти М., Бургуэн Л. и др. (2013). Фолли Ф (ред.). «Иммунопозитивность к макроизоформам гистонов H2A1 отмечает гепатоцеллюлярную карциному, связанную со стеатозом» . ПЛОС ОДИН . 8 (1): e54458. Бибкод : 2013PLoSO...854458R . дои : 10.1371/journal.pone.0054458 . ПМЦ 3553099 . ПМИД 23372727 .
- ^ Роперо С., Фрага М.Ф., Баллестар Е., Хамелин Р., Ямамото Х., Буа-Шорне М. и др. (май 2006 г.). «Усекательная мутация HDAC2 при раке человека придает устойчивость к ингибированию деацетилазы гистонов». Природная генетика . 38 (5): 566–569. дои : 10.1038/ng1773 . ПМИД 16642021 . S2CID 9073684 .
- ^ Верхельст, Сигрид; Ван Пуйвельде, Барт; Виллемс, Сандер; Далед, Саймон; Корнелис, Сенн; Корвелейн, Лаура; Виллемс, Эвуд; Дефорс, Дитер; ДеКлерк, Лаура; Дэненс, Мартен (24 января 2022 г.). «Крупномасштабный скрининг гистонов на основе масс-спектрометрии для оценки эпигенетической токсичности для развития» . Научные отчеты . 12 (1): 1256. Бибкод : 2022НатСР..12.1256В . дои : 10.1038/s41598-022-05268-x . hdl : 1854/LU-8735551 . ISSN 2045-2322 . ПМЦ 8786925 . ПМИД 35075221 .
- ^ ван Аттикум Х., Гассер С.М. (май 2009 г.). «Перекрестные помехи между модификациями гистонов во время реакции на повреждение ДНК». Тенденции в клеточной биологии . 19 (5): 207–217. дои : 10.1016/j.tcb.2009.03.001 . ПМИД 19342239 .
- ^ Фридман Р.К., Фарх К.К., Бердж CB, Бартель Д.П. (январь 2009 г.). «Большинство мРНК млекопитающих являются консервативными мишенями микроРНК» . Геномные исследования . 19 (1): 92–105. дои : 10.1101/гр.082701.108 . ПМЦ 2612969 . ПМИД 18955434 .
- ^ Сайто Ю., Лян Г., Эггер Дж., Фридман Дж. М., Чуанг Дж. К., Кутзи Г. А., Джонс П. А. (июнь 2006 г.). «Специфическая активация микроРНК-127 с подавлением протоонкогена BCL6 препаратами, модифицирующими хроматин, в раковых клетках человека» . Раковая клетка . 9 (6): 435–443. дои : 10.1016/j.ccr.2006.04.020 . ПМИД 16766263 .
- ^ Луджамбио А., Роперо С., Баллестар Е., Фрага М.Ф., Серрато С., Сетьен Ф. и др. (февраль 2007 г.). «Генетическое разоблачение эпигенетически заглушенной микроРНК в раковых клетках человека» . Исследования рака . 67 (4): 1424–1429. дои : 10.1158/0008-5472.CAN-06-4218 . ПМИД 17308079 .
- ^ Jump up to: а б Сото-Рейес Э., Гонсалес-Барриос Р., Сиснерос-Соберанис Ф., Эррера-Гепферт Р., Перес В., Канту Д. и др. (январь 2012 г.). «Нарушение CTCF в локусе миР-125b1 при гинекологическом раке» . БМК Рак . / 1471-2407-12-40 дои : 10.1186 . ПМЦ 3297514 . ПМИД 22277129 .
- ^ Врба Л., Муньос-Родригес Х.Л., Стампфер М.Р., Футшер Б.В. (2013). «Промоторы генов микроРНК часто являются мишенью аберрантного метилирования ДНК при раке молочной железы человека» . ПЛОС ОДИН . 8 (1): e54398. Бибкод : 2013PLoSO...854398V . дои : 10.1371/journal.pone.0054398 . ПМЦ 3547033 . ПМИД 23342147 .
- ^ Jump up to: а б Ван Ю.П., Лэй Цюй (май 2018 г.). «Метаболическая перекодировка эпигенетики рака» . Раковые коммуникации . 38 (1): 25. дои : 10.1186/s40880-018-0302-3 . ПМЦ 5993135 . ПМИД 29784032 .
- ^ Кастан М.Б. (апрель 2008 г.). «Реакция на повреждение ДНК: механизмы и роль в заболеваниях человека: лекция на премию Мемориала ГСГ Клоуза 2007 г.» . Молекулярные исследования рака . 6 (4): 517–524. дои : 10.1158/1541-7786.MCR-08-0020 . ПМИД 18403632 .
- ^ Jump up to: а б Бернштейн С., Прасад А.Р., Нфонсам В., Бернштейн Х. (2013). «Глава 16: Повреждение ДНК, восстановление ДНК и рак». В Чэнь С. (ред.). Новые направления исследований в области репарации ДНК . Совет директоров – Книги по запросу. п. 413. ИСБН 978-953-51-1114-6 .
- ^ Jump up to: а б О'Хаган Х.М., Мохаммад Х.П., Бэйлин С.Б. (август 2008 г.). «Двухнитевые разрывы могут инициировать молчание генов и SIRT1-зависимое начало метилирования ДНК на экзогенном промоторном острове CpG» . ПЛОС Генетика . 4 (8): е1000155. дои : 10.1371/journal.pgen.1000155 . ПМЦ 2491723 . ПМИД 18704159 .
- ^ Jump up to: а б Куоццо С., Порчеллини А., Ангризано Т., Морано А., Ли Б., Ди Пардо А. и др. (июль 2007 г.). «Повреждение ДНК, репарация, направленная на гомологию, и метилирование ДНК» . ПЛОС Генетика . 3 (7): е110. дои : 10.1371/journal.pgen.0030110 . ЧВК 1913100 . ПМИД 17616978 .
- ^ Джасперсон К.В., Туохи Т.М., Некласон Д.В., Берт Р.В. (июнь 2010 г.). «Наследственный и семейный рак толстой кишки» . Гастроэнтерология . 138 (6): 2044–2058. дои : 10.1053/j.gastro.2010.01.054 . ПМК 3057468 . ПМИД 20420945 .
- ^ Ван Г, Матур Р., Ху Х, Чжан Х, Лу Х (сентябрь 2011 г.). «Реакция микроРНК на повреждение ДНК» . Тенденции биохимических наук . 36 (9): 478–484. дои : 10.1016/j.tibs.2011.06.002 . ПМЦ 3532742 . ПМИД 21741842 .
- ^ Тесситоре А., Чиччарелли Г., Дель Веккио Ф., Гаджиано А., Верцелла Д., Фискьетти М. и др. (2014). «МикроРНК в сети повреждения/восстановления ДНК и раке» . Международный журнал геномики . 2014 : 820248. doi : 10.1155/2014/820248 . ПМЦ 3926391 . ПМИД 24616890 .
- ^ Jump up to: а б Шнекенбургер М., Дидерих М. (март 2012 г.). «Эпигенетика открывает новые горизонты в профилактике колоректального рака» . Текущие отчеты о колоректальном раке . 8 (1): 66–81. дои : 10.1007/s11888-011-0116-z . ПМК 3277709 . ПМИД 22389639 .
- ^ Jump up to: а б Валери Н., Гаспарини П., Фаббри М., Бракони С., Веронезе А., Ловат Ф. и др. (апрель 2010 г.). «Модуляция репарации ошибочных спариваний и стабильности генома с помощью миР-155» . Труды Национальной академии наук Соединенных Штатов Америки . 107 (15): 6982–6987. Бибкод : 2010PNAS..107.6982V . дои : 10.1073/pnas.1002472107 . JSTOR 25665289 . ПМЦ 2872463 . ПМИД 20351277 .
- ^ Jump up to: а б Трунингер К., Менигатти М., Луз Дж., Рассел А., Хайдер Р., Гебберс Дж.О. и др. (май 2005 г.). «Иммуногистохимический анализ выявляет высокую частоту дефектов PMS2 при колоректальном раке» . Гастроэнтерология . 128 (5): 1160–1171. дои : 10.1053/j.gastro.2005.01.056 . ПМИД 15887099 .
- ^ Jump up to: а б с Чжан В., Чжан Дж., Ходли К., Кушваха Д., Рамакришнан В., Ли С. и др. (июнь 2012 г.). «миР-181d: прогнозирующий биомаркер глиобластомы, который подавляет экспрессию MGMT» . Нейроонкология . 14 (6): 712–719. дои : 10.1093/neuonc/nos089 . ПМЦ 3367855 . ПМИД 22570426 .
- ^ Шпигль-Крейнекер С., Пиркер С., Филипитс М., Лётч Д., Бухройтнер Дж., Пихлер Дж. и др. (январь 2010 г.). «Экспрессия белка O6-метилгуанин-ДНК-метилтрансферазы в опухолевых клетках предсказывает результат терапии темозоломидом у пациентов с глиобластомой» . Нейроонкология . 12 (1): 28–36. дои : 10.1093/neuonc/nop003 . ПМЦ 2940563 . ПМИД 20150365 .
- ^ Jump up to: а б Палмьери Д., Д'Анджело Д., Валентино Т., Де Мартино И., Ферраро А., Виринкс А. и др. (август 2012 г.). «Понижающая регуляция микроРНК, нацеленных на HMGA, играет решающую роль в онкогенезе гипофиза человека» . Онкоген . 31 (34): 3857–3865. дои : 10.1038/onc.2011.557 . ПМИД 22139073 .
- ^ Сгарра Р., Рустиги А., Тессари М.А., Ди Бернардо Дж., Альтамура С., Фуско А. и др. (сентябрь 2004 г.). «Ядерные фосфопротеины HMGA и их связь со структурой хроматина и раком». Письма ФЭБС . 574 (1–3): 1–8. дои : 10.1016/j.febslet.2004.08.013 . ПМИД 15358530 . S2CID 28903539 .
- ^ Сюй Ю, Самтер Т.Ф., Бхаттачарья Р., Тесфай А., Фукс Э.Дж., Вуд Л.Дж. и др. (май 2004 г.). «Онкоген HMG-I вызывает высоко проникающие агрессивные лимфоидные злокачественные новообразования у трансгенных мышей и сверхэкспрессируется при лейкемии человека». Исследования рака . 64 (10): 3371–3375. дои : 10.1158/0008-5472.CAN-04-0044 . ПМИД 15150086 . S2CID 34111491 .
- ^ Бальдасарре Дж., Баттиста С., Беллетти Б., Такур С., Пентималли Ф., Трапассо Ф. и др. (апрель 2003 г.). «Негативная регуляция экспрессии гена BRCA1 белками HMGA1 объясняет снижение уровня белка BRCA1 при спорадической карциноме молочной железы» . Молекулярная и клеточная биология . 23 (7): 2225–2238. дои : 10.1128/MCB.23.7.2225-2238.2003 . ПМК 150734 . ПМИД 12640109 .
- ^ Боррманн Л., Шванбек Р., Хейдук Т., Зеебек Б., Рогалла П., Буллердик Дж., Вишневски Дж. Р. (декабрь 2003 г.). «Белок группы А2 с высокой подвижностью и его производные связывают конкретную область промотора гена репарации ДНК ERCC1 и модулируют его активность» . Исследования нуклеиновых кислот . 31 (23): 6841–6851. дои : 10.1093/nar/gkg884 . ПМК 290254 . ПМИД 14627817 .
- ^ Jump up to: а б с д Фациста А., Нгуен Х., Льюис С., Прасад А.Р., Рэмси Л., Зайтлин Б. и др. (апрель 2012 г.). «Недостаточная экспрессия ферментов репарации ДНК на ранней стадии развития спорадического рака толстой кишки» . Целостность генома . 3 (1): 3. дои : 10.1186/2041-9414-3-3 . ПМК 3351028 . ПМИД 22494821 .
- ^ Малумбрес М (2013). «МиРНК и рак: взгляд эпигенетики» . Молекулярные аспекты медицины . 34 (4): 863–874. дои : 10.1016/j.mam.2012.06.005 . ПМК 5791883 . ПМИД 22771542 .
- ^ Сампат Д., Лю С., Васан К., Сульда М., Пудувалли В.К., Вирда В.Г., Китинг М.Дж. (февраль 2012 г.). «Гистоновые деацетилазы опосредуют подавление миР-15а, миР-16 и миР-29b при хроническом лимфоцитарном лейкозе» . Кровь . 119 (5): 1162–1172. doi : 10.1182/blood-2011-05-351510 . ПМК 3277352 . ПМИД 22096249 .
- ↑ Гены восстановления ДНК человека , 15 апреля 2014 г., Онкологический центр доктора медицины Андерсона, Техасский университет.
- ^ Кришнан К., Степто А.Л., Мартин Х.К., Вани С., Нонес К., Уодделл Н. и др. (февраль 2013 г.). «МикроРНК-182-5p нацелена на сеть генов, участвующих в репарации ДНК» . РНК . 19 (2): 230–242. дои : 10.1261/rna.034926.112 . ПМК 3543090 . ПМИД 23249749 .
- ^ Чайсаингмонгкол Дж., Попанда О., Варта Р., Дайкхофф Г., Херпель Е., Гейзельхарт Л. и др. (декабрь 2012 г.). «Эпигенетический скрининг генов репарации ДНК человека выявляет аберрантное метилирование промотора NEIL1 при плоскоклеточном раке головы и шеи» . Онкоген . 31 (49): 5108–5116. дои : 10.1038/onc.2011.660 . ПМИД 22286769 .
- ^ Лян Л., Дэн Л., Чен Ю., Ли Г.К., Шао С., Тишфилд Дж.А. (сентябрь 2005 г.). «Модуляция соединения концов ДНК ядерными белками» . Журнал биологической химии . 280 (36): 31442–31449. дои : 10.1074/jbc.M503776200 . ПМИД 16012167 .
- ^ Сингх П., Ян М., Дай Х., Ю Д., Хуан К., Тан В. и др. (ноябрь 2008 г.). «Сверхэкспрессия и гипометилирование гена лоскутной эндонуклеазы 1 при раке молочной железы и других видах рака» . Молекулярные исследования рака . 6 (11): 1710–1717. дои : 10.1158/1541-7786.MCR-08-0269 . ПМЦ 2948671 . ПМИД 19010819 .
- ^ Лам Дж.С., Селигсон Д.Б., Ю Х., Ли А., Иева М., Пантак А.Дж. и др. (август 2006 г.). «Эндонуклеаза лоскута 1 сверхэкспрессируется при раке простаты и связана с высоким показателем Глисона». БЖУ Интернешнл . 98 (2): 445–451. дои : 10.1111/j.1464-410X.2006.06224.x . ПМИД 16879693 . S2CID 22165252 .
- ^ Ким Дж.М., Сон ХИ, Юн С.И., О Дж.Х., Ян Джо, Ким Дж.Х. и др. (январь 2005 г.). «Идентификация генов, связанных с раком желудка, с использованием микроматрицы кДНК, содержащей новые метки экспрессируемых последовательностей, экспрессируемых в клетках рака желудка» . Клинические исследования рака . 11 (2 ч. 1): 473–482. дои : 10.1158/1078-0432.473.11.2 . ПМИД 15701830 .
- ^ Ван К., Се С., Чен Д. (май 2014 г.). «Эндонуклеаза лоскута 1 является многообещающим биомаркером-кандидатом при раке желудка и участвует в пролиферации и апоптозе клеток» . Международный журнал молекулярной медицины . 33 (5): 1268–1274. дои : 10.3892/ijmm.2014.1682 . ПМИД 24590400 .
- ^ Краузе А., Комбаре В., Яконо И., Лакруа Б., Компаньон С., Бержерон С. и др. (июль 2005 г.). «Полногеномный анализ экспрессии генов в нейробластомах, обнаруженных с помощью массового скрининга» (PDF) . Письма о раке . 225 (1): 111–120. дои : 10.1016/j.canlet.2004.10.035 . ПМИД 15922863 . S2CID 44644467 .
- ^ Якобузио-Донахью К.А., Майтра А., Олсен М., Лоу А.В., ван Хик Н.Т., Рости С. и др. (апрель 2003 г.). «Исследование глобальных закономерностей экспрессии генов при аденокарциноме поджелудочной железы с использованием микрочипов кДНК» . Американский журнал патологии . 162 (4): 1151–1162. дои : 10.1016/S0002-9440(10) 63911-9 ПМЦ 1851213 . ПМИД 12651607 .
- ^ Сато М., Жирар Л., Секине И., Сунага Н., Рамирес Р.Д., Камибаяши К., Минна Дж.Д. (октябрь 2003 г.). «Повышенная экспрессия и отсутствие мутаций гена эндонуклеазы Flap (FEN1) при раке легких человека». Онкоген . 22 (46): 7243–7246. дои : 10.1038/sj.onc.1206977 . ПМИД 14562054 . S2CID 22443138 .
- ^ Би ФФ, Ли Д, Ян Кью (2013). «Гипометилирование сайтов связывания транскрипционных факторов ETS и усиление экспрессии PARP1 при раке эндометрия» . БиоМед Исследования Интернэшнл . 2013 : 946268. doi : 10.1155/2013/946268 . ПМЦ 3666359 . ПМИД 23762867 .
- ^ Ли Д., Би Ф.Ф., Цао Дж.М., Цао С., Ли С.И., Лю Б., Ян Ц. (январь 2014 г.). «Регуляция транскрипции поли (АДФ-рибоза) полимеразы 1: новое перекрестное взаимодействие между модификацией гистонов H3K9ac и гипометилированием мотива ETS1 при раке яичников с мутацией BRCA1» . Онкотаргет . 5 (1): 291–297. дои : 10.18632/oncotarget.1549 . ПМК 3960209 . ПМИД 24448423 .
- ^ Би ФФ, Ли Д, Ян Ц (февраль 2013 г.). «Гипометилирование промотора, особенно вокруг мотива, специфичного для трансформации E26, и повышенная экспрессия поли(АДФ-рибозы) полимеразы 1 при серозном раке яичников с мутацией BRCA» . БМК Рак . 13:90 . дои : 10.1186/1471-2407-13-90 . ПМК 3599366 . ПМИД 23442605 .
- ^ Нарайанан Л., Фритцелл Дж.А., Бейкер С.М., Лискай Р.М., Глейзер П.М. (апрель 1997 г.). «Повышенные уровни мутаций во многих тканях мышей с дефицитом гена репарации несоответствия ДНК Pms2» . Труды Национальной академии наук Соединенных Штатов Америки . 94 (7): 3122–3127. Бибкод : 1997PNAS...94.3122N . дои : 10.1073/pnas.94.7.3122 . JSTOR 41786 . ЧВК 20332 . ПМИД 9096356 .
- ^ Хеган Д.С., Нараянан Л., Жирик Ф.Р., Эдельманн В., Лискай Р.М., Глейзер П.М. (декабрь 2006 г.). «Различные модели генетической нестабильности у мышей с дефицитом генов репарации ошибочного спаривания Pms2, Mlh1, Msh2, Msh3 и Msh6» . Канцерогенез . 27 (12): 2402–2408. doi : 10.1093/carcin/bgl079 . ПМК 2612936 . ПМИД 16728433 .
- ^ Тутт А.Н., ван Остром КТ, Росс ГМ, ван Стиг Х., Эшворт А. (март 2002 г.). «Нарушение Brca2 увеличивает частоту спонтанных мутаций in vivo: синергизм с ионизирующим излучением» . Отчеты ЭМБО . 3 (3): 255–260. дои : 10.1093/embo-reports/kvf037 . ПМК 1084010 . ПМИД 11850397 .
- ^ Хэлфорд С., Роуэн А., Сойер Э., Талбот I, Томлинсон I (июнь 2005 г.). «О (6)-метилгуанин метилтрансфераза при колоректальном раке: обнаружение мутаций, потеря экспрессии и слабая связь с переходами G: C> A: T» . Гут . 54 (6): 797–802. дои : 10.1136/gut.2004.059535 . ПМК 1774551 . ПМИД 15888787 .
- ^ Шен Л., Кондо Ю., Рознер Г.Л., Сяо Л., Эрнандес Н.С., Вилайтонг Дж. и др. (сентябрь 2005 г.). «Метилирование промотора MGMT и дефект поля при спорадическом колоректальном раке» . Журнал Национального института рака . 97 (18): 1330–1338. дои : 10.1093/jnci/dji275 . ПМИД 16174854 .
- ^ Jump up to: а б Ли К.Х., Ли Дж.С., Нам Дж.Х., Чой С., Ли MC, Парк К.С. и др. (октябрь 2011 г.). «Статус метилирования промотора генов hMLH1, hMSH2 и MGMT при колоректальном раке, связанном с последовательностью аденома-карцинома». Архив хирургии Лангенбека . 396 (7): 1017–1026. дои : 10.1007/s00423-011-0812-9 . ПМИД 21706233 . S2CID 8069716 .
- ^ Сврчек М., Бухард О., Колас С., Куле Ф., Дюмон С., Массауди И. и др. (ноябрь 2010 г.). «Толерантность к метилированию из-за дефекта поля O6-метилгуанин-ДНК-метилтрансферазы (MGMT) в слизистой оболочке толстой кишки: начальный этап в развитии колоректального рака с дефицитом репарации несоответствия». Гут . 59 (11): 1516–1526. дои : 10.1136/gut.2009.194787 . ПМИД 20947886 . S2CID 206950452 .
- ^ Палущак Ю., Мисяк П., Вежбицка М., Возняк А., Баер-Дубовска В. (февраль 2011 г.). «Частое гиперметилирование DAPK, RARbeta, MGMT, RASSF1A и FHIT при плоскоклеточном раке гортани и прилегающей нормальной слизистой оболочке». Оральная онкология . 47 (2): 104–107. doi : 10.1016/j.oraloncology.2010.11.006 . ПМИД 21147548 .
- ^ Цзо С., Чжан Х., Спенсер Х.Дж., Вурал Е., Суен Дж.Ю., Шичман С.А. и др. (октябрь 2009 г.). «Повышенная микросателлитная нестабильность и эпигенетическая инактивация гена hMLH1 при плоскоклеточном раке головы и шеи». Отоларингология – хирургия головы и шеи . 141 (4): 484–490. дои : 10.1016/j.otohns.2009.07.007 . ПМИД 19786217 . S2CID 8357370 .
- ^ Тауфик Х.М., Эль-Максуд Н.М., Хак Б.Х., Эль-Щербини Ю.М. (2011). «Плоскоклеточный рак головы и шеи: иммуногистохимия восстановления несоответствия и гиперметилирование промотора гена hMLH1». Американский журнал отоларингологии . 32 (6): 528–536. дои : 10.1016/j.amjoto.2010.11.005 . ПМИД 21353335 .
- ^ Цзоу XP, Чжан Б, Чжан XQ, Чен М, Цао Дж, Лю WJ (ноябрь 2009 г.). «Промоторное гиперметилирование нескольких генов при ранней аденокарциноме желудка и предраковых поражениях». Патология человека . 40 (11): 1534–1542. дои : 10.1016/j.humpath.2009.01.029 . ПМИД 19695681 .
- ^ Вани М., Афроз Д., Махдуми М., Хамид И., Вани Б., Бхат Дж. и др. (2012). «Статус метилирования промотора гена репарации ДНК (hMLH1) у пациентов с карциномой желудка в Кашмирской долине» . Азиатско-Тихоокеанский журнал профилактики рака . 13 (8): 4177–4181. дои : 10.7314/APJCP.2012.13.8.4177 . ПМИД 23098428 .
- ^ Jump up to: а б Раза Ю., Ахмед А., Хан А., Чишти А.А., Ахтер С.С., Мубарак М. и др. (май 2020 г.). «Helicobacter pylori серьезно снижает экспрессию белков репарации ДНК PMS2 и ERCC1 при гастрите и раке желудка» . Восстановление ДНК . 89 : 102836. doi : 10.1016/j.dnarep.2020.102836 . ПМИД 32143126 . S2CID 212622031 .
- ^ Агарвал А., Полинени Р., Хусейн З., Вигода И., Бхагат Т.Д., Бхаттачарья С. и др. (2012). «Роль эпигенетических изменений в патогенезе пищевода Барретта и аденокарциномы пищевода» . Международный журнал клинической и экспериментальной патологии . 5 (5): 382–396. ПМК 3396065 . ПМИД 22808291 .
- ^ Туна М., Амос С.И. (ноябрь 2013 г.). «Геномное секвенирование при раке» . Письма о раке . 340 (2): 161–170. дои : 10.1016/j.canlet.2012.11.004 . ПМЦ 3622788 . ПМИД 23178448 .
- ^ Роуч Дж.К., Глусман Дж., Смит А.Ф., Хафф К.Д., Хабли Р., Шеннон П.Т. и др. (апрель 2010 г.). «Анализ генетического наследования в семейном квартете методом полногеномного секвенирования» . Наука . 328 (5978): 636–639. Бибкод : 2010Sci...328..636R . дои : 10.1126/science.1186802 . ПМК 3037280 . ПМИД 20220176 .
- ^ Кэмпбелл С.Д., Чонг Дж.К., Малиг М., Ко А., Дюмон Б.Л., Хан Л. и др. (ноябрь 2012 г.). «Оценка частоты мутаций у человека с использованием аутозиготности в популяции основателей» . Природная генетика . 44 (11): 1277–1281. дои : 10.1038/ng.2418 . ПМЦ 3483378 . ПМИД 23001126 .
- ^ Кейтли П.Д. (февраль 2012 г.). «Скорость и последствия новых мутаций у человека» . Генетика . 190 (2): 295–304. дои : 10.1534/genetics.111.134668 . ПМК 3276617 . ПМИД 22345605 .
- ^ Йе К., Бикман М., Ламейер Э.В., Чжан Ю., Моед М.Х., ван ден Аккер Э.Б. и др. (декабрь 2013 г.). «Старение как ускоренное накопление соматических вариантов: полногеномное секвенирование пар столетних и монозиготных близнецов среднего возраста» . Исследования близнецов и генетика человека . 16 (6): 1026–1032. дои : 10.1017/thg.2013.73 . ПМИД 24182360 .
- ^ Шанбхаг Н.М., Рафальска-Меткалф И.Ю., Балане-Боливар С., Яницки С.М., Гринберг Р.А. (июнь 2010 г.). «АТМ-зависимый хроматин меняет молчание транскрипции в цис-цис-системе на двухцепочечные разрывы ДНК» . Клетка . 141 (6): 970–981. дои : 10.1016/j.cell.2010.04.038 . ПМК 2920610 . ПМИД 20550933 .
- ^ Морано А., Ангрисано Т., Руссо Г., Ланди Р., Пезоне А., Бартоллино С. и др. (январь 2014 г.). «Направленное метилирование ДНК путем гомологии-направленного восстановления в клетках млекопитающих. Транскрипция изменяет форму метилирования восстановленного гена» . Исследования нуклеиновых кислот . 42 (2): 804–821. дои : 10.1093/нар/gkt920 . ПМК 3902918 . ПМИД 24137009 .
- ^ Фернандес А.Ф., Асенов Ю., Мартин-Суберо Дж.И., Балинт Б., Зиберт Р., Танигучи Х. и др. (февраль 2012 г.). «Отпечаток метилирования ДНК 1628 образцов человека» . Геномные исследования . 22 (2): 407–419. дои : 10.1101/гр.119867.110 . ПМК 3266047 . ПМИД 21613409 .
- ^ Епископ Дж.Б., Витт К.Л., Слоан Р.А. (декабрь 1997 г.). «Генетическая токсичность тератогенов человека» . Мутационные исследования . 396 (1–2): 9–43. дои : 10.1016/S0027-5107(97)00173-5 . ПМИД 9434858 .
- ^ Гурвич Н., Берман М.Г., Виттнер Б.С., Джентльмен Р.К., Кляйн П.С., Грин Дж.Б. (июль 2005 г.). «Связь тератогенеза, индуцированного вальпроатом, с ингибированием деацетилазы гистонов in vivo» . Журнал ФАСЭБ . 19 (9): 1166–1168. doi : 10.1096/fj.04-3425fje . ПМИД 15901671 . S2CID 25874971 .
- ^ Смителлс Д. (ноябрь 1998 г.). «Вызывает ли талидомид врожденные дефекты второго поколения?». Безопасность лекарств . 19 (5): 339–341. дои : 10.2165/00002018-199819050-00001 . ПМИД 9825947 . S2CID 9014237 .
- ^ Фридлер Г. (декабрь 1996 г.). «Отцовское воздействие: влияние на репродуктивные результаты и развитие. Обзор» . Фармакология, биохимия и поведение . 55 (4): 691–700. дои : 10.1016/S0091-3057(96)00286-9 . ПМИД 8981601 . S2CID 2260876 .
- ^ «Вкладыш в упаковку Видазы (азацитидин для инъекционной суспензии)» (PDF) . Компания Фармион . Управление по контролю за продуктами и лекарствами США. 18 мая 2004 г. Архивировано из оригинала (PDF) 29 мая 2004 г.
- ^ Цицерон Т.Дж., Адамс М.Л., Джордано А., Миллер Б.Т., О'Коннор Л., Нок Б. (март 1991 г.). «Влияние воздействия морфина в подростковом возрасте на половое созревание самцов крыс и развитие их потомства». Журнал фармакологии и экспериментальной терапии . 256 (3): 1086–1093. ПМИД 2005573 .
- ^ Ньюболд Р.Р., Падилья-Бэнкс Э., Джефферсон В.Н. (июнь 2006 г.). «Неблагоприятное воздействие модельного экологического эстрогена диэтилстильбестрола передается последующим поколениям» . Эндокринология . 147 (6 Доп.): С11–С17. дои : 10.1210/en.2005-1164 . ПМИД 16690809 .
- ^ Jump up to: а б Катияр С.К., Сингх Т., Прасад Р., Сун К., Вайд М. (сентябрь 2012 г.). «Эпигенетические изменения в канцерогенезе кожи, индуцированном ультрафиолетовым излучением: взаимодействие биоактивных пищевых компонентов с эпигенетическими мишенями» . Фотохимия и фотобиология . 88 (5): 1066–1074. дои : 10.1111/j.1751-1097.2011.01020.x . ПМЦ 3288155 . ПМИД 22017262 .
- ^ Оруджи Э., Утикал Дж. (ноябрь 2018 г.). «Эпигенетическая борьба со злокачественной меланомой: метилирование лизина гистонов» . Клиническая эпигенетика . 10 (1): 145. дои : 10.1186/s13148-018-0583-z . ПМК 6249913 . ПМИД 30466474 .
- ^ Jump up to: а б Коллинз CC, Волик С.В., Лапук А.В., Ван Ю, Гут П.В., Ву С. и др. (март 2012 г.). «Секвенирование рака простаты нового поколения у пациента выявляет дефицит метилтиоаденозинфосфорилазы, пригодной для использования мишени опухоли» . Молекулярная терапия рака . 11 (3): 775–783. дои : 10.1158/1535-7163.MCT-11-0826 . ПМЦ 3691697 . ПМИД 22252602 .
- ^ Jump up to: а б Ли Л.К., Кэрролл П.Р., Дахия Р. (январь 2005 г.). «Эпигенетические изменения при раке простаты: значение для диагностики и лечения». Журнал Национального института рака . 97 (2): 103–115. дои : 10.1093/jnci/dji010 . ПМИД 15657340 .
- ^ Jump up to: а б Гурель Б., Ивата Т., Ко К.М., Егнасубраманиан С., Нельсон В.Г., Де Марзо А.М. (ноябрь 2008 г.). «Молекулярные изменения при раке простаты как диагностические, прогностические и терапевтические цели» . Достижения анатомической патологии . 15 (6): 319–331. дои : 10.1097/PAP.0b013e31818a5c19 . ПМК 3214657 . ПМИД 18948763 .
- ^ Орниш Д., Магбануа М.Дж., Вайднер Г., Вайнберг В., Кемп С., Грин С. и др. (июнь 2008 г.). «Изменения в экспрессии генов простаты у мужчин, подвергающихся интенсивному питанию и изменению образа жизни» . Труды Национальной академии наук Соединенных Штатов Америки . 105 (24): 8369–8374. Бибкод : 2008PNAS..105.8369O . дои : 10.1073/pnas.0803080105 . ПМК 2430265 . ПМИД 18559852 .
- ^ Jump up to: а б с д и ж г час Сан С., Реймерс Л.Л., Берк Р.Д. (апрель 2011 г.). «Метилирование CpG-сайтов генома HPV16 связано с предраком и раком шейки матки» . Гинекологическая онкология . 121 (1): 59–63. дои : 10.1016/j.ygyno.2011.01.013 . ПМК 3062667 . ПМИД 21306759 .
- ^ Мандал СС (апрель 2010 г.). «Лейкемия смешанного происхождения: универсальный игрок в эпигенетике и заболеваниях человека» . Журнал ФЭБС . 277 (8): 1789. doi : 10.1111/j.1742-4658.2010.07605.x . ПМИД 20236314 . S2CID 37705117 .
- ^ Неббиозо А, Тамбаро Ф.П., Делл'Аверсана С, Альтуччи Л (июнь 2018 г.). Действительно GM (ред.). «Эпигенетика рака: движение вперед» . ПЛОС Генетика . 14 (6): e1007362. дои : 10.1371/journal.pgen.1007362 . ПМЦ 5991666 . ПМИД 29879107 .
- ^ Мясник Дж. (март 2001 г.). «Электромагнитные поля могут вызвать лейкемию у детей». Ланцет . 357 (9258): 777. doi : 10.1016/S0140-6736(05)71207-1 . S2CID 54400632 .
- ^ «Саркома мягких тканей» . Национальный институт рака . Национальные институты здравоохранения, Министерство здравоохранения и социальных служб США. Январь 1980 года.
- ^ Беннани-Баити ИМ (декабрь 2011 г.). «Эпигенетические и эпигеномные механизмы формируют патогенез саркомы и других мезенхимальных опухолей». Эпигеномика . 3 (6): 715–732. дои : 10.2217/эпи.11.93 . ПМИД 22126291 .
- ^ Рихтер Г.Х., Плем С., Фасан А., Рёсслер С., Унланд Р., Беннани-Баити И.М. и др. (март 2009 г.). «EZH2 является медиатором роста опухоли и метастазирования, обусловленного EWS/FLI1, блокируя эндотелиальную и нейроэктодермальную дифференцировку» . Труды Национальной академии наук Соединенных Штатов Америки . 106 (13): 5324–5329. Бибкод : 2009PNAS..106.5324R . дои : 10.1073/pnas.0810759106 . ПМЦ 2656557 . ПМИД 19289832 .
- ^ Беннани-Баити IM, Мачадо I, Лломбарт-Бош А, Ковар Х (август 2012 г.). «Лизин-специфическая деметилаза 1 (LSD1/KDM1A/AOF2/BHC110) экспрессируется и является эпигенетической мишенью лекарственного средства при хондросаркоме, саркоме Юинга, остеосаркоме и рабдомиосаркоме». Патология человека . 43 (8): 1300–1307. дои : 10.1016/j.humpath.2011.10.010 . ПМИД 22245111 .
- ^ Тэм, Кит В.; Чжан, Вэй; Сох, Джуничи; Стастный, Виктор; Чен, Мин; Сунь, Хан; Четверг, Келси; Риос, Джонатан Дж.; Ян, Ченчен; Марконетт, Кристал Н.; Селамат, Сухайда. А.; Лэрд-Оффринга, Ите А.; Тагучи, Аюму; Ханаш, Самир; Позор, Дэвид (01 ноября 2013 г.). «Механизмы инактивации CDKN2A/p16 и их связь с воздействием дыма и молекулярными особенностями при немелкоклеточном раке легких» . Журнал торакальной онкологии . 8 (11): 1378–1388. дои : 10.1097/JTO.0b013e3182a46c0c . ISSN 1556-0864 . ПМЦ 3951422 . ПМИД 24077454 .
- ^ Хонг, Руню; Лю, Венке; Феньё, Дэвид (2021). «Прогнозирование и визуализация мутации STK11 на слайдах гистопатологии аденокарциномы легких с использованием глубокого обучения» . дои : 10.1101/2020.02.20.956557 . Проверено 16 марта 2023 г.
- ^ Ахмад, Аамир (2015). Рак легких и персонализированная медицина: новые методы лечения и клиническое лечение . Международное издательство Спрингер. ISBN 978-3-319-24932-2 . OCLC 1113687835 .
- ^ Ивакава, Рейка; Коно, Анами, Ёичи; Судзуки, Кендзи; Мисима, Кадзухико; Таширо, Фумио, Дзюн (15 июня 2008 г.) . Гомозиготные делеции p16 с клинико-патологическими характеристиками . » при / и EGFR / аденокарциноме KRAS мутациями легких p53 ПМИД 18559592 . S2CID 17931210 .
- ^ Фонтан, JW; Генденинг, Дж. М.; Флорес, Дж. Ф. (1 сентября 1994 г.). «Характеристика гена p16 у мышей: доказательства существования большого семейства генов» . Американский журнал генетики человека . 55 (Приложение 3). ОСТИ 133832 .
- ^ Эстеллер М., Герман Дж.Г. (январь 2004 г.). «Создание мутаций, но обеспечение химиочувствительности: роль O6-метилгуанин-ДНК-метилтрансферазы при раке человека». Онкоген . 23 (1): 1–8. дои : 10.1038/sj.onc.1207316 . ПМИД 14712205 . S2CID 38574543 .
- ^ Эстеллер М., Гарсиа-Фонсиллас Дж., Андион Э., Гудман С.Н., Идальго О.Ф., Ванаклоха В. и др. (ноябрь 2000 г.). «Инактивация гена репарации ДНК MGMT и клинический ответ глиом на алкилирующие агенты». Медицинский журнал Новой Англии . 343 (19): 1350–1354. дои : 10.1056/NEJM200011093431901 . hdl : 2445/176306 . ПМИД 11070098 . S2CID 40303322 .
- ^ Хеги М.Е., Дизеренс А.С., Горлия Т., Хаму М.Ф., де Триболе Н., Веллер М. и др. (март 2005 г.). «Подавление гена MGMT и польза темозоломида при глиобластоме» (PDF) . Медицинский журнал Новой Англии . 352 (10): 997–1003. doi : 10.1056/NEJMoa043331 . ПМИД 15758010 .
- ^ Эстеллер М., Гайдано Г., Гудман С.Н., Загонель В., Капелло Д., Ботто Б. и др. (январь 2002 г.). «Гиперметилирование гена репарации ДНК О (6)-метилгуанин ДНК-метилтрансферазы и выживаемость пациентов с диффузной крупноклеточной В-клеточной лимфомой» . Журнал Национального института рака . 94 (1): 26–32. дои : 10.1093/jnci/94.1.26 . ПМИД 11773279 .
- ^ Гласспул Р.М., Теодоридис Дж.М., Браун Р.М. (апрель 2006 г.). «Эпигенетика как механизм полигенной клинической лекарственной устойчивости» . Британский журнал рака . 94 (8): 1087–1092. дои : 10.1038/sj.bjc.6603024 . ПМК 2361257 . ПМИД 16495912 .
- ^ Брок М.В., Хукер С.М., Ота-Мачида Е., Хан Ю., Го М., Эймс С. и др. (март 2008 г.). «Маркеры метилирования ДНК и ранний рецидив рака легких I стадии» . Медицинский журнал Новой Англии . 358 (11): 1118–1128. doi : 10.1056/NEJMoa0706550 . ПМИД 18337602 . S2CID 18279123 .
- ^ Jump up to: а б Иглесиас-Линарес А., Яньес-Вико Р.М., Гонсалес-Молес М.А. (май 2010 г.). «Потенциальная роль ингибиторов HDAC в терапии рака: понимание плоскоклеточного рака полости рта». Оральная онкология . 46 (5): 323–329. doi : 10.1016/j.oraloncology.2010.01.009 . ПМИД 20207580 .
- ^ Ван Л.Г., Цзяо Дж.В. (сентябрь 2010 г.). «Химиопрофилактическая активность фенетилизотиоцианата при раке простаты посредством эпигенетической регуляции (обзор)» . Международный журнал онкологии . 37 (3): 533–539. дои : 10.3892/ijo_00000702 . ПМИД 20664922 .
- ^ Герардини Л., Шарма А., Капобьянко Э., Синти С. (27 мая 2016 г.). «Нацеливание на рак с помощью эпилекарств: взгляд на точную медицину». Современная фармацевтическая биотехнология . 17 (10): 856–865. дои : 10.2174/1381612822666160527154757 . ПМИД 27229488 .
- ^ Спаннхофф А., Сиппл В., Юнг М. (январь 2009 г.). «Лечение рака будущего: ингибиторы гистоновых метилтрансфераз». Международный журнал биохимии и клеточной биологии . 41 (1): 4–11. doi : 10.1016/j.biocel.2008.07.024 . ПМИД 18773966 .
- ^ Гарсия-Манеро Дж., Штольц М.Л., Уорд М.Р., Кантарджян Х., Шарма С. (сентябрь 2008 г.). «Пилотное фармакокинетическое исследование перорального азацитидина». Лейкемия . 22 (9): 1680–1684. дои : 10.1038/leu.2008.145 . ПМИД 18548103 . S2CID 19854416 .
- ^ Гарсия-Манеро Дж. (ноябрь 2008 г.). «Деметилирующие агенты при миелоидных новообразованиях» . Современное мнение в онкологии . 20 (6): 705–710. дои : 10.1097/CCO.0b013e328313699c . ПМЦ 3873866 . ПМИД 18841054 .
- ^ Ариби А., Бортакур Г., Раванди Ф., Шан Дж., Дэвиссон Дж., Кортес Дж., Кантарджян Х. (февраль 2007 г.). «Активность децитабина, гипометилирующего агента, при хроническом миеломоноцитарном лейкозе» . Рак . 109 (4): 713–717. дои : 10.1002/cncr.22457 . ПМИД 17219444 .
- ^ Де Падуа Силва Л., де Лима М., Кантарджян Х., Фадерл С., Кебриаи П., Гиральт С. и др. (июнь 2009 г.). «Возможность алло-СКТ после гипометилирующей терапии децитабином при миелодиспластическом синдроме». Трансплантация костного мозга . 43 (11): 839–843. дои : 10.1038/bmt.2008.400 . ПМИД 19151791 . S2CID 42034181 .
- ^ Хамбах Л., Линг К.В., Пул Дж., Агай З., Блокланд Э., Танке Х.Дж. и др. (март 2009 г.). «Гипометилирующие препараты превращают HA-1-отрицательные солидные опухоли в мишени для иммунотерапии на основе стволовых клеток» . Кровь . 113 (12): 2715–2722. дои : 10.1182/blood-2008-05-158956 . ПМИД 19096014 . S2CID 206871351 .
- ^ Фено П., Муфтий Г.Дж., Хеллстрем-Линдберг Е., Сантини В., Финелли С., Джагунидис А. и др. (март 2009 г.). «Эффективность азацитидина по сравнению с эффективностью традиционных схем лечения при лечении миелодиспластических синдромов повышенного риска: рандомизированное открытое исследование III фазы» . «Ланцет». Онкология . 10 (3): 223–232. дои : 10.1016/S1470-2045(09)70003-8 . ПМК 4086808 . ПМИД 19230772 .
- ^ Дувик М., Талпур Р., Ни Х, Чжан С., Хазарика П., Келли С. и др. (январь 2007 г.). «Фаза 2 исследования перорального вориностата (субероиланилид гидроксамовой кислоты, SAHA) при рефрактерной кожной Т-клеточной лимфоме (CTCL)» . Кровь . 109 (1): 31–39. дои : 10.1182/кровь-2006-06-025999 . ПМК 1785068 . ПМИД 16960145 .
- ^ Олсен Э.А., Ким Ю.Х., Кузель Т.М., Пачеко Т.Р., Фосс Ф.М., Паркер С. и др. (июль 2007 г.). «Многоцентровое исследование фазы IIb вориностата у пациентов с персистирующей, прогрессирующей или резистентной к лечению кожной Т-клеточной лимфомой». Журнал клинической онкологии . 25 (21): 3109–3115. дои : 10.1200/JCO.2006.10.2434 . ПМИД 17577020 . S2CID 19558322 .
- ^ Кэмерон Э.Э., Бахман К.Э., Мёханен С., Герман Дж.Г., Байлин С.Б. (январь 1999 г.). «Синергия деметилирования и ингибирования деацетилазы гистонов при повторной экспрессии генов, молчащих при раке». Природная генетика . 21 (1): 103–107. дои : 10.1038/5047 . ПМИД 9916800 . S2CID 25070861 .
- ^ «Применение нового препарата: Панобиностат» (PDF) . Центр оценки и исследования лекарственных средств . Управление по контролю за продуктами и лекарствами США. 14 января 2015 г.
- ^ Дауден Дж., Хонг В., Парри Р.В., Пайк Р.А., Уорд С.Г. (апрель 2010 г.). «На пути к разработке мощных и селективных бисубстратных ингибиторов протеинаргининметилтрансфераз». Письма по биоорганической и медицинской химии . 20 (7): 2103–2105. дои : 10.1016/j.bmcl.2010.02.069 . ПМИД 20219369 .
- ^ Гальвез А.Ф., Чен Н., Макасиб Дж., де Люмен Б.О. (15 октября 2001 г.). «Химиопрофилактическое свойство соевого пептида (луназина), который связывается с деацетилированными гистонами и ингибирует ацетилирование» . Исследования рака . 61 .
- ^ Браун, Роберт; Стратди, Гордон (1 апреля 2002 г.). «Эпигеномика и эпигенетическая терапия рака» . Тенденции молекулярной медицины . 8 (4): С43–С48. дои : 10.1016/S1471-4914(02)02314-6 . ISSN 1471-4914 . ПМИД 11927287 .
- ^ Эггер, Герда; Лян, Ганнин; Апарисио, Ана; Джонс, Питер А. (май 2004 г.). «Эпигенетика в заболеваниях человека и перспективы эпигенетической терапии» . Природа . 429 (6990): 457–463. Бибкод : 2004Natur.429..457E . дои : 10.1038/nature02625 . ISSN 1476-4687 . ПМИД 15164071 . S2CID 4424126 .
- ^ Сон, Сан-Хён; Хан, Саэ-Вон; Банг, Юн-Джюэ (01 декабря 2011 г.). «Эпигенетическая терапия рака» . Наркотики . 71 (18): 2391–2403. дои : 10.2165/11596690-000000000-00000 . ISSN 1179-1950 . ПМИД 22141383 . S2CID 35003553 .
- ^ Салим, Мохаммед (май 2015 г.). «Эпигенетическая терапия рака» . Пак. Дж. Фарм. Наука . 28 (3): 1023–1032.
- ^ Jump up to: а б aacrjournals.org https://aacrjournals.org/clincancerres/article/13/6/1634/195851/DNA-Mmethylation-as-a-Therapeutic-Target-in-Cancer . Проверено 22 октября 2022 г.
{{cite web}}: Отсутствует или пусто|title=( помощь ) - ^ Кантарджян, Акоп; Исса, Жан-Пьер Ж.; Розенфельд, Крейг С.; Беннетт, Джон М.; Альбитар, Махер; ДиПерсио, Джон; Климат, Вирджиния; Слэк, Джеймс; Кастро, Чарльз; Раванди, Фархад; Хелмер, Ричард; Шен, Ланлан; Наймер, Стивен Д.; Ливитт, Ричард; Рэйс, Азра (15 апреля 2006 г.). «Децитабин улучшает результаты лечения пациентов с миелодиспластическими синдромами: результаты рандомизированного исследования III фазы» . Рак 106 (8): 1794–1803. дои : 10.1002/cncr.21792 . ISSN 0008-543X . ПМИД 16532500 . S2CID 9556660 .