Наследственный раковый синдром

Наследственный раковый синдром ( синдром семейного/семейного рака , синдром наследственного рака , синдром предрасположенности к раку , синдром рака и т. д.) — генетическое заболевание , при котором наследственные генетические мутации в одном или нескольких генах предрасполагают больных к развитию рака и могут также вызывают раннее начало этих видов рака. Наследственные раковые синдромы часто демонстрируют не только высокий пожизненный риск развития рака, но и развитие множественных независимых первичных опухолей . [1]
Многие из этих синдромов вызваны мутациями в генах-супрессорах опухолей , генах, которые участвуют в защите клетки от превращения в раковую. Другими генами, которые могут быть затронуты, являются репарации ДНК гены , онкогены и гены, участвующие в образовании кровеносных сосудов ( ангиогенез ). [2] Распространенными примерами наследственных раковых синдромов являются наследственный синдром рака молочной железы и яичников и наследственный неполипозный рак толстой кишки (синдром Линча). [3] [4]
Фон
[ редактировать ]Наследственные раковые синдромы лежат в основе от 5 до 10% всех случаев рака, и существует более 50 идентифицируемых наследственных форм рака. [5] Научное понимание синдромов предрасположенности к раку активно расширяется: обнаруживаются дополнительные синдромы, [6] лежащая в основе биология становится яснее, а генетическое тестирование улучшает выявление, лечение и профилактику раковых синдромов. [7] Учитывая распространенность рака молочной железы и толстой кишки, наиболее широко распространенные синдромы включают наследственный синдром рака молочной железы и яичников и наследственный неполипозный рак толстой кишки (синдром Линча). [6]
Некоторые редкие виды рака тесно связаны с синдромами наследственной предрасположенности к раку. Генетическое тестирование следует рассмотреть при адренокортикальной карциноме ; карциноидные опухоли ; диффузный рак желудка ; маточная труба/первичный рак брюшины ; лейомиосаркома ; медуллярный рак щитовидной железы ; параганглиома /феохромоцитома; почечно-клеточная карцинома хромофобной, гибридной онкоцитарной или онкоцитомной гистологии; сальная карцинома ; и опухоли полового канатика с кольцевидными канальцами. [6] Врачи первичной медико-санитарной помощи могут выявить людей, подверженных риску наследственного ракового синдрома. [8]
Генетика рака
[ редактировать ]
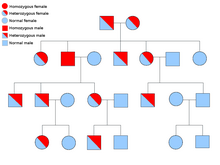
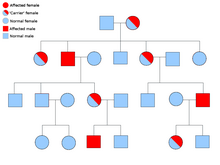
Две копии каждого гена присутствуют во всех клетках организма, и каждая из них называется аллелью . Большинство раковых синдромов передаются по менделевскому аутосомно-доминантному типу. В этих случаях для того, чтобы у человека была предрасположенность к раку, должен присутствовать только один дефектный аллель. Лица с одним нормальным аллелем и одним дефектным аллелем называются гетерозиготными . Гетерозиготный человек и человек с двумя нормальными аллелями ( гомозиготный ) будут иметь 50% вероятность рождения больного ребенка. [9] Мутация унаследованного гена известна как мутация зародышевой линии , а дальнейшая мутация нормального аллеля приводит к развитию рака. Это известно как гипотеза двух ударов Кнудсона , согласно которой первое попадание гена является наследственной мутацией, а второе совпадение происходит в более позднем возрасте. [2] Поскольку необходимо мутировать только один аллель (по сравнению с обоими при так называемом «спорадическом раке»), у человека более высокий шанс развития рака, чем у населения в целом. [10]
Реже синдромы могут передаваться по аутосомно-рецессивному признаку. Оба аллеля гена должны быть мутированы при аутосомно-рецессивных заболеваниях, чтобы у человека появилась предрасположенность к раку. Человек с двумя рецессивными аллелями называется гомозиготным рецессивным . Оба родителя должны иметь хотя бы один дефектный аллель, чтобы ребенок был гомозиготным рецессивным. Если у обоих родителей есть один мутантный аллель и один нормальный аллель ( гетерозиготный ), то у них есть 25% шанс произвести на свет гомозиготного рецессивного ребенка (имеет предрасположенность), 50% шанс произвести гетерозиготного ребенка (носитель дефектного гена) и 25% шанс. вероятность рождения ребенка с двумя нормальными аллелями. [9]
Примерами аутосомно-доминантных раковых синдромов являются аутоиммунный лимфопролиферативный синдром (синдром Канале-Смита), синдром Беквита-Видемана (хотя 85% случаев являются спорадическими), [ нужна ссылка ] Синдром Бирта-Хогга-Дюбе , синдром Карни , семейная хордома , синдром Каудена , синдром диспластического невуса с семейной меланомой , семейный аденоматозный полипоз , наследственный синдром рака молочной железы и яичников , наследственный диффузный рак желудка (HDGC), Наследственный неполипозный колоректальный рак (синдром Линча) , синдром Хауэла-Эванса рака пищевода с тилозом , синдром ювенильного полипоза , синдром Ли-Фраумени , множественная эндокринная неоплазия типа 1/2, множественный остеохондроматоз , нейрофиброматоз типа 1/2, синдром невоидной базальноклеточной карциномы (синдром Горлина), Пейтца- синдром Егерса , семейный рак предстательной железы , наследственный лейомиоматозный почечно-клеточный рак (LRCC), наследственный папиллярный почечно-клеточный рак , наследственный параганглиомы синдром -феохромоцитомы, ретинобластома , туберозный склероз , болезнь фон Гиппеля-Линдау и опухоль Вильмса . [11]
Примерами синдромов аутосомно-рецессивного рака являются атаксия-телеангиэктазия , синдром Блума , анемия Фанкони , MUTYH-ассоциированный полипоз, синдром Ротмунда-Томсона , синдром Вернера и пигментная ксеродерма . [11]
Примеры
[ редактировать ]Хотя раковые синдромы демонстрируют повышенный риск развития рака, риск варьируется. Для некоторых из этих заболеваний рак не является основным признаком. [ нужна ссылка ]
Анемия Фанкони
[ редактировать ]Анемия Фанкони — заболевание с широким клиническим спектром, включающим раннее начало и повышенный риск развития рака; недостаточность костного мозга ; и врожденные аномалии . Наиболее выраженные проявления этого расстройства связаны с кроветворением (выработкой крови костным мозгом ); к ним относятся апластическая анемия , миелодиспластический синдром и острый миелоидный лейкоз . Опухоли печени и плоскоклеточный рак пищевода представляют , ротоглотки и язычка собой солидные опухоли, обычно связанные с ФА. К врожденным аномалиям относятся: аномалии скелета (особенно затрагивающие руки), пятна цвета кофе с молоком и гипопигментация . На сегодняшний день известны гены, вызывающие ФА: FANCA , FANCB , FANCC , FANCD2 , FANCE , FANCF , FANCG , FANCI , FANCJ , FANCL , FANCM , FANCN , FANCO , FANCP и BRCA2 (ранее известный как FANCD1). Наследование этого синдрома преимущественно аутосомно-рецессивное , но FANCB может наследоваться от материнской или отцовской Х-хромосомы ( х-сцепленное рецессивное наследование ). Путь FA участвует в репарации ДНК, когда две цепи ДНК неправильно соединены ( межцепочечные сшивки ). Многие пути для этого координируются путем FA, включая эксцизионную репарацию нуклеотидов , синтез транслейкоза и гомологичную рекомбинацию . [12] [13] [14] [15] [16]
Семейный аденоматозный полипоз
[ редактировать ]Семейный аденоматозный полипоз (САП) — это аутосомно-доминантный синдром, который значительно увеличивает риск колоректального рака . Это заболевание встречается примерно у 1 из 8000 человек, и его пенетрантность составляет примерно 100% . появляются сотни и тысячи доброкачественных аденом У человека с этим заболеванием в толстой кишке , которые в большинстве случаев перерастают в рак. Другие опухоли, частота которых увеличивается, включают; остеомы , аденомы и карциномы надпочечников , опухоли щитовидной железы и десмоидные опухоли . Причиной этого расстройства является мутировавший ген APC , который участвует в регуляции β-катенина . Неисправный APC вызывает накопление β-катенина в клетках и активацию факторов транскрипции, участвующих в клеток пролиферации , миграции , дифференцировке и апоптозе (запрограммированной гибели клеток). [17] [18] [19]
Наследственный рак молочной железы и яичников
[ редактировать ]Наследственный синдром рака молочной железы и яичников — аутосомно-доминантное генетическое заболевание, вызванное генетическими мутациями генов BRCA1 и BRCA2 . У женщин это заболевание в первую очередь увеличивает риск рака молочной железы и яичников , но также увеличивает риск рака фаллопиевой трубы и папиллярного серозного рака брюшины. риск рака простаты У мужчин повышен . Другими видами рака, которые непоследовательно связаны с этим синдромом, являются рак поджелудочной железы , рак молочной железы у мужчин , колоректальный рак и рак матки и шейки матки . На генетические мутации приходится примерно 7% и 14% случаев рака молочной железы и яичников соответственно, а на BRCA1 и BRCA2 приходится 80% этих случаев. BRCA1 и BRCA2 являются генами-супрессорами опухолей , участвующими в поддержании и восстановлении ДНК, что, в свою очередь, приводит к нестабильности генома. Мутации в этих генах приводят к дальнейшему повреждению ДНК, что может привести к раку. [20] [21]
Наследственный неполипозный рак толстой кишки
[ редактировать ]Наследственный неполипозный рак толстой кишки , также известный как синдром Линча, представляет собой аутосомно-доминантный раковый синдром, который увеличивает риск развития колоректального рака. Это вызвано генетическими мутациями в генах восстановления несоответствия ДНК (MMR), особенно MLH1 , MSH2 , MSH6 и PMS2 . Помимо колоректального рака, увеличивается частота возникновения многих других видов рака. К ним относятся; рак эндометрия , рак желудка , рак яичников , рак тонкой кишки и рак поджелудочной железы . Наследственный неполипозный рак толстой кишки также связан с ранним началом колоректального рака. Гены MMR участвуют в восстановлении ДНК, когда основания каждой цепи ДНК не совпадают. Дефектные гены MMR допускают непрерывные мутации вставки и делеции в областях ДНК, известных как микросателлиты . Эти короткие повторяющиеся последовательности ДНК становятся нестабильными, что приводит к состоянию микросателлитной нестабильности (MSI). Мутированные микросателлиты часто обнаруживаются в генах, участвующих в инициировании и прогрессировании опухолей, а MSI может повысить выживаемость клеток, что приводит к раку. [4] [22] [23] [24]
Большинство случаев семейной параганглиомы вызваны мутациями в генах субъединицы сукцинатдегидрогеназы (сукцинат:убихиноноксидоредуктаза) ( SDHD , SDHAF2 , SDHC , SDHB ).
PGL-1 связан с мутацией SDHD, и у большинства людей с параганглиомой PGL-1 страдают отцы, а не матери. PGL1 и PGL2 являются аутосомно-доминантными с импринтингом . PGL-4 связан с мутацией SDHB и связан с более высоким риском феохромоцитомы, а также почечно-клеточного рака и немедуллярного рака щитовидной железы. [25]
Синдром Ли-Фраумени
[ редактировать ]Синдром Ли-Фраумени — это аутосомно-доминантный синдром, вызываемый преимущественно мутациями гена TP53 , который значительно увеличивает риск многих видов рака, а также тесно связан с ранним началом этих видов рака. Рак, связанный с этим расстройством, включает; саркомы мягких тканей (часто обнаруживаются в детском возрасте), остеосаркома , рак молочной железы , рак головного мозга , лейкемия и адренокортикальная карцинома . У людей с синдромом Ли-Фраумени часто наблюдаются множественные независимые первичные раковые заболевания. Причина широкого клинического спектра этого расстройства может быть связана с мутациями других генов, которые модифицируют заболевание. Белок p53, продуцируемый геном TP53 , участвует в остановке клеточного цикла , восстановлении ДНК и апоптозе . Дефектный р53 может быть не в состоянии должным образом выполнять эти процессы, что может быть причиной образования опухоли. Поскольку только 60-80% людей с этим расстройством имеют обнаруживаемые мутации в TP53 , другие мутации в пути p53 могут быть вовлечены в синдром Ли-Фраумени. [26] [27] [28] [29] Лицам с СЛФ необходим интенсивный скрининг на протяжении всей жизни для раннего выявления рака. [30] Для получения дополнительной информации см. Синдром Ли-Фраумени .
MUTYH-ассоциированный полипоз
[ редактировать ]MUTYH-ассоциированный полипоз разделяет большинство своих клинических особенностей с FAP; разница в том, что это аутосомно-рецессивное заболевание, вызванное мутациями в MUTYH гене репарации ДНК . Опухолями повышенного риска при этом заболевании являются колоректальный рак, аденомы желудка и аденомы двенадцатиперстной кишки. [17] [31]

Синдром невоидной базальноклеточной карциномы
[ редактировать ]Синдром невоидной базальноклеточной карциномы , также известный как синдром Горлина, представляет собой аутосомно-доминантный раковый синдром, при котором риск базальноклеточной карциномы очень высок. Заболевание характеризуется базальноклеточными невусами , кератоцистами челюстей и аномалиями скелета. Оценки распространенности синдрома невоидной базальноклеточной карциномы варьируются, но составляют примерно 1 на 60 000. Наличие базальноклеточной карциномы гораздо чаще встречается у белых, чем у чернокожих людей; 80% и 38% соответственно. Одонтогенные кератоцисты обнаруживаются примерно у 75% людей с этим заболеванием и часто возникают в раннем возрасте. Наиболее распространенные скелетные аномалии возникают в области головы и лица, но часто поражаются и другие области, например, грудная клетка . Причинная генетическая мутация этого заболевания происходит в гене PTCH , а продукт PTCH является супрессором опухоли, участвующим в передаче сигналов в клетках . Хотя точная роль этого белка в синдроме невоидной базальноклеточной карциномы неизвестна, он участвует в сигнальном пути ежа. , который, как известно, контролирует клеток . рост и развитие [32] [33]
Болезнь фон Гиппеля-Линдау
[ редактировать ]Болезнь фон Гиппеля-Линдау — редкое аутосомно-доминантное генетическое заболевание, которое предрасполагает людей к доброкачественным и злокачественным опухолям. Наиболее распространенными опухолями при болезни Фон Хиппеля-Линдау являются гемангиобластомы центральной нервной системы и сетчатки, светлоклеточный рак почки, феохромоцитомы, нейроэндокринные опухоли поджелудочной железы, кисты поджелудочной железы, опухоли эндолимфатического мешка и папиллярные цистаденомы придатка яичка. [34] [35] Болезнь фон Хиппеля-Линдау возникает в результате мутации гена-супрессора опухоли фон Хиппеля-Линдау на хромосоме 3p25.3. [36]
Пигментная ксеродерма
[ редактировать ]Пигментная ксеродерма — аутосомно-рецессивное заболевание, характеризующееся чувствительностью к ультрафиолетовому (УФ) свету , значительно повышенным риском солнечных ожогов и повышенным риском рака кожи . Риск рака кожи более чем в 10 000 раз выше, чем у здоровых людей, и включает в себя многие виды рака кожи, включая меланому и немеланомный рак кожи. Кроме того, подверженные воздействию солнца участки языка, губ и глаз имеют повышенный риск развития рака. Пигментная ксеродерма может быть связана с другими видами рака внутренних органов и доброкачественными опухолями. [ нужна ссылка ] Помимо рака, некоторые генетические мутации , вызывающие пигментную ксеродермию, связаны с нейродегенерацией . Пигментная ксеродерма может быть вызвана генетическими мутациями в 8 генах, которые продуцируют следующие ферменты : XPA , XPB , XPC , XPD , XPE , XPF , XPG и Pol η . XPA-XPF — это ферменты эксцизионной репарации нуклеотидов , которые восстанавливают ДНК, поврежденную ультрафиолетовым светом, а дефектные белки способствуют накоплению мутаций, вызванных ультрафиолетовым светом. Pol η представляет собой полимеразу , которая представляет собой фермент, участвующий в репликации ДНК. Существует множество полимераз, но pol η — это фермент, который реплицирует ДНК, поврежденную ультрафиолетовым светом. Мутации в этом гене приводят к образованию дефектного фермента pol η, который не может реплицировать ДНК при повреждении ультрафиолетовым светом. Лица с мутациями этого гена имеют подмножество XP; XP-вариант заболевания. [37] [38]
Дефекты репарации ДНК и повышенный риск рака
[ редактировать ]Многие раковые синдромы возникают из-за наследственного нарушения способности репарации ДНК . [ нужна ссылка ] Когда в гене репарации ДНК присутствует унаследованная мутация , ген репарации либо не экспрессируется, либо экспрессируется в измененной форме. Тогда функция репарации, скорее всего, будет нарушена, и, как следствие, повреждения ДНК будут иметь тенденцию к накоплению. Такие повреждения ДНК могут вызвать ошибки во время синтеза ДНК, приводящие к мутациям, некоторые из которых могут привести к раку. Мутации репарации ДНК зародышевой линии, повышающие риск развития рака, перечислены в таблице.
| Ген репарации ДНК | Белок | Затронутые пути восстановления* | Рак с повышенным риском |
|---|---|---|---|
| атаксия телеангиэктазия мутировала | Банкомат | Различные мутации в ATM снижают HRR , SSA или NHEJ. [39] | лейкемия, лимфома, молочная железа [39] [40] |
| Синдром Блума | БЛМ ( геликаза ) | ЧРР [41] | лейкемия, лимфома, толстая кишка, молочная железа, кожа, легкие, слуховой проход, язык, пищевод, желудок, миндалины, гортань, матка [42] |
| рак молочной железы 1 и 2 | BRCA1 BRCA2 | HRR двухцепочечных разрывов и разрывов дочерних цепей [43] | грудь, яичник [44] |
| Гены анемии Фанкони FANCA,B,C,D1,D2,E,F,G,I,J,L,M,N,O,P | ФАНКА и др. | HRR и TLS [45] | лейкемия, опухоли печени, солидные опухоли многих областей [46] |
| наследственного неполипозного колоректального рака Гены MSH2 MSH6 MLH1 PMS2 | МШ2 МШ6 МЛХ1 ПМС2 | ММР [47] | колоректальный, эндометрий, яичники, желудочно-кишечный тракт (желудок и тонкий кишечник, поджелудочная железа, желчевыводящие пути), мочевыводящие пути, головной мозг (глиобластомы) и кожа (кератоакантомы и сальные аденомы) [48] |
| синдрома Ли-Фраумени Ген TP53 | P53 | Непосредственная роль в HRR, BER, NER и в реакции на повреждение ДНК. [49] для этих путей, а также для NHEJ и MMR [50] | саркомы, рак молочной железы, опухоли головного мозга и адренокортикальные карциномы [51] |
| МРЭ11А | МРЭ11 | HRR и NHEJ [52] | грудь [53] |
| МУТЫХ | MUTYH гликозилаза | BER A в паре с 8-оксо-dG [54] | колоректальный рак, рак двенадцатиперстной кишки, яичников, мочевого пузыря и кожи [55] |
| Синдром поломки Неймегена | НБС (НБН) | НХЭЖ [56] | лимфоидный рак [56] |
| НТХЛ1 | НТХЛ1 | BER для Tg, FapyG, 5-hC, 5-hU в дцДНК [57] | Рак толстой кишки , рак эндометрия , рак двенадцатиперстной кишки , базальноклеточный рак [58] |
| РЕККЛ4 | РЕКК4 | Хеликаза, вероятно, активна при HRR [59] | базальноклеточный рак, плоскоклеточный рак, внутриэпидермальный рак [60] |
| синдрома Вернера Ген WRN | Синдром Вернера АТФ-зависимая геликаза | HRR, NHEJ, длинный патч BER [61] | саркома мягких тканей, колоректальный рак, кожа, щитовидная железа, поджелудочная железа [62] |
| пигментной ксеродерма Гены XPA , XPB , XPD , XPF , XPG | XPA XPB XPD XPF XPG | Транскрипционно-связанный NER восстанавливает транскрибируемые цепи транскрипционно активных генов. [63] | рак кожи (меланома и немеланома) [63] |
| пигментной ксеродерма Гены XPC , XPE ( DDB2 ) | ХПК, ХПЭ | Глобальный геномный NER восстанавливает повреждения как в транскрибируемой, так и в нетранскрибируемой ДНК. [37] [64] | рак кожи (меланома и немеланома) [37] [64] |
| XPV (также называемый полимеразой H) | DNA polymerase eta (Pol η) | Синтез транслезий (TLS) [65] | рак кожи (базальноклеточный, плоскоклеточный, меланома) [65] |
- Акронимами путей репарации ДНК являются гомологичная рекомбинационная репарация HRR, подпуть SSA HRR , негомологичное соединение концов BER NHEJ, эксцизионная репарация оснований TLS , трансочаговый синтез NER , эксцизионная репарация нуклеотидов MMR , репарация несоответствия .
Генетический скрининг
[ редактировать ]Генетическое тестирование можно использовать для выявления мутировавших генов или хромосом , которые передаются из поколения в поколение. Люди с положительным результатом теста на наличие генетической мутации не обязательно обречены на развитие рака, связанного с мутацией, однако они обладают повышенным риском развития рака по сравнению с населением в целом. Людям рекомендуется пройти генетический тест, если их семейный медицинский анамнез включает в себя: несколько членов семьи, больных раком, кто-то из членов семьи, заболевший раком в особенно молодом возрасте или принадлежащий к определенной этнической группе . [7]

Процесс генетического скрининга представляет собой простую и неинвазивную процедуру. Однако прежде чем гены будут проверены на наличие мутаций, пациент обычно должен обратиться к врачу и пройти индивидуальную консультацию , на которой обсуждается как личный, так и семейный анамнез рака. Затем медицинский работник может оценить вероятность наличия у пациента мутации и провести процедуру генетического скрининга. [66] Важно, чтобы эта консультация состоялась, поскольку она гарантирует, что человек даст информированное согласие на участие в генетическом тестировании, будет осведомлен и понимает этапы, преимущества и ограничения процедуры, а также будет лучше осведомлен о последствиях результатов проверки слуха. [67] Тест можно провести с использованием жидкостей организма или клеток пациента, включая; кровь (чаще всего), слюна, околоплодные воды и даже клетки внутренней полости рта, полученные из буккального мазка . Затем этот материал отправляется в специализированную генетическую лабораторию, где его изучают технические специалисты, результаты анализов отправляются обратно поставщику медицинских услуг, запросившему анализ, и результаты обсуждаются с пациентом. [7]
Прямое тестирование на потребителе можно провести без участия медицинского работника, но оно не рекомендуется, поскольку потребитель теряет возможность обсудить свое решение с образованным профессионалом. [68] По данным Национальной медицинской библиотеки США, генетическое тестирование в Америке стоит в диапазоне от 100 до 2000 долларов в зависимости от типа и сложности теста. [69]
Профилактические действия
[ редактировать ]Генетическое тестирование важно, поскольку, если тест окажется положительным, они будут лучше осведомлены о своем личном здоровье и здоровье ближайших членов семьи. [70] С помощью и советом медицинского специалиста они могут предпринять шаги для снижения повышенного риска развития рака посредством:
- Регулярные упражнения
- Здоровое, сбалансированное питание
- Поддержание здорового веса
- Не курить
- Как оставаться в безопасности под вредными солнечными лучами [71]
Существуют и другие формы профилактических действий. Примером наследственного рака молочной железы и рака яичников может быть хирургическое вмешательство: гистерэктомия — это удаление всей или части матки , тогда как мастэктомия — это удаление груди ( двойная мастэктомия означает, что обе груди удаляются), это часто может увеличить продолжительность их жизни на несколько лет . [72] Еще одной профилактической мерой является регулярное обследование и обследование на предмет выявления рака. Если у человека синдром Линча , ему следует регулярно проходить колоноскопию, чтобы проверить, есть ли какие-либо изменения в клетках, выстилающих стенку кишечника. Регулярные осмотры связаны с дополнительными 7 годами продолжительности жизни в среднем для человека с синдромом Линча. синдром. Это связано с тем, что раннее выявление означает, что правильные профилактические действия и операция могут быть проведены быстрее. [73] Регулярный скрининг молочных желез также рекомендуется женщинам с диагнозом мутации BRCA . Недавние исследования показывают, что мужчины с повышенным риском развития рака простаты из-за мутаций BRCA могут снизить этот риск, принимая аспирин . [74] Аспирин чрезвычайно полезен для снижения распространенности рака; однако для достижения какого-либо эффекта его необходимо принимать регулярно в течение как минимум пятилетнего периода. [75]
Распространенность генетических мутаций в разных этнических группах
[ редактировать ]Часто генетические мутации более распространены в определенных этнических группах, это связано с тем, что раса может отследить своих предков до одного географического местоположения, мутировавшие гены затем передаются от предков из поколения в поколение, поэтому некоторые этнические группы более восприимчивы к мутациям, что увеличивает их шансы на развитие рака [61]. Как упоминалось выше, это может быть полезно, поскольку помогает медицинским работникам оценить риск возникновения мутации у пациента до того, как он пройдет тестирование. [66] синдрома Вернера составляет 1 на 200 000 живорождений в США, но в Японии он поражает людей в 1 на 20 000–40 000 случаев. Распространенность [76] У 1 из 40 евреев-ашкенази есть мутация BRCA, что резко контрастирует с общей численностью населения в Соединенных Штатах, где ею страдает 1 из 400 человек. Евреи-ашкенази подвергаются высокому риску развития наследственного рака молочной железы и яичников, поэтому им рекомендуется пройти как генетическое тестирование, чтобы определить, есть ли у них мутация, так и регулярный скрининг на рак. [77]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Аллгайер, Хайке; Редер, Хельга; Фульда, Симона (2009). Наследственные опухоли: от генов к клиническим последствиям . Вайнхайм: Wiley-VCH. ISBN 9783527320288 .
- ^ Перейти обратно: а б Ходжсон С. (январь 2008 г.). «Механизмы наследственной предрасположенности к раку» . J Чжэцзянский университет, бакалавр наук . 9 (1): 1–4. дои : 10.1631/jzus.B073001 . ПМК 2170461 . ПМИД 18196605 .
- ^ Кларк А.С., Домчек С.М. (апрель 2011 г.). «Клиническое лечение наследственных синдромов рака молочной железы». J Биол неоплазия молочной железы . 16 (1): 17–25. дои : 10.1007/s10911-011-9200-x . ПМИД 21360002 . S2CID 21417924 .
- ^ Перейти обратно: а б Линч Х.Т., Линч П.М., Ланспа С.Дж., Снайдер К.Л., Линч Дж.Ф., Боланд С.Р. (июль 2009 г.). «Обзор синдрома Линча: история, молекулярная генетика, скрининг, дифференциальный диагноз и судебно-медицинские последствия» . Клин. Жене . 76 (1): 1–18. дои : 10.1111/j.1399-0004.2009.01230.x . ПМК 2846640 . ПМИД 19659756 .
- ^ «Генетика» . Национальный институт рака . 22 апреля 2015 г. Проверено 20 февраля 2018 г.
- ^ Перейти обратно: а б с Бэнкс, КС; Молин, Джей-Джей; Марвин, ML; Ньюлин, AC; Фогель, К.Дж. (март 2013 г.). «10 редких опухолей, требующих направления к генетику». Семейный рак . 12 (1): 1–18. дои : 10.1007/s10689-012-9584-9 . ПМИД 23377869 . S2CID 14426194 .
- ^ Перейти обратно: а б с «Генетическое тестирование на наследственные раковые синдромы» . Национальный институт рака . 22 апреля 2013 г. Проверено 19 февраля 2018 г.
- ^ Корде, Лариса А.; Гадалла, Шахиназ М. (2 мая 2017 г.). «Оценка риска рака для врача первичной медико-санитарной помощи» . Первичный уход . 36 (3): 471–488. дои : 10.1016/j.pop.2009.04.006 . ПМЦ 2713871 . ПМИД 19616151 .
- ^ Перейти обратно: а б Андерсон, Синди Лу; Кэри А. Браун (2007). Патофизиология: функциональные изменения в здоровье человека . Хагерствон, доктор медицины: Липпинкотт Уильямс и Уилкинс. ISBN 978-0-7817-6250-2 .
- ^ Сабувала, доктор Хаким К. (10 апреля 2022 г.). Что такое раковый синдром или семейный раковый синдром? Краткий обзор . Доктор Хаким Сабувала.
- ^ Перейти обратно: а б Линдор Н.М., Грин М.Х. (июль 1998 г.). «Краткий справочник семейных раковых синдромов. Программа семейного рака Мэйо» . Журнал Национального института рака . 90 (14): 1039–71. дои : 10.1093/jnci/90.14.1039 . ПМИД 9672254 .
- ^ Молдавский Г.Л., Д'Андреа А.Д. (2009). «Как путь анемии Фанкони охраняет геном» . Анну. Преподобный Жене . 43 : 223–49. doi : 10.1146/annurev-genet-102108-134222 . ПМЦ 2830711 . ПМИД 19686080 .
- ^ Тишковитц, доктор медицинских наук, Ходжсон С.В. (январь 2003 г.). «Анемия Фанкони» . Журнал медицинской генетики . 40 (1): 1–10. дои : 10.1136/jmg.40.1.1 . ПМЦ 1735271 . ПМИД 12525534 .
- ^ Ки Ю, Д'Андреа А.Д. (ноябрь 2012 г.). «Молекулярный патогенез и клиническое лечение анемии Фанкони» . Журнал клинических исследований . 122 (11): 3799–806. дои : 10.1172/JCI58321 . ПМЦ 3484428 . ПМИД 23114602 .
- ^ Коттеманн MC, Смогожевска А (январь 2013 г.). «Анемия Фанкони и восстановление перекрестных связей ДНК Уотсона и Крика» . Природа . 493 (7432): 356–63. Бибкод : 2013Natur.493..356K . дои : 10.1038/nature11863 . ПМК 3700363 . ПМИД 23325218 .
- ^ Су X, Хуан Дж (сентябрь 2011 г.). «Путь анемии Фанкони и восстановление межцепочечных поперечных связей ДНК» . Белковая клетка . 2 (9): 704–11. дои : 10.1007/s13238-011-1098-y . ПМЦ 4875268 . ПМИД 21948210 .
- ^ Перейти обратно: а б Халф Э, Беркович Д, Розен П (2009). «Семейный аденоматозный полипоз» . Orphanet J Редкий дис . 4:22 . дои : 10.1186/1750-1172-4-22 . ПМЦ 2772987 . ПМИД 19822006 .
- ^ Галиацатос П., Фоулкс В.Д. (февраль 2006 г.). «Семейный аденоматозный полипоз». Американский журнал гастроэнтерологии . 101 (2): 385–98. дои : 10.1111/j.1572-0241.2006.00375.x . ПМИД 16454848 . S2CID 8516051 .
- ^ Макрэ Ф., Дю Сарт Д., Насиулас С. (2009). «Семейный аденоматозный полипоз». Best Pract Res Clin Гастроэнтерол . 23 (2): 197–207. дои : 10.1016/j.bpg.2009.02.010 . ПМИД 19414146 .
- ^ Петручелли Н., Дейли М.Б., Фельдман Г.Л. (май 2010 г.). «Наследственный рак молочной железы и яичников вследствие мутаций BRCA1 и BRCA2» . Жене. Мед . 12 (5): 245–59. дои : 10.1097/GIM.0b013e3181d38f2f . ПМИД 20216074 .
- ^ Смит ЕС (2012). «Обзор наследственного синдрома рака молочной железы и яичников». J Акушерство и женское здоровье . 57 (6): 577–84. дои : 10.1111/j.1542-2011.2012.00199.x . ПМИД 23050669 .
- ^ Дрешер К.М., Шарма П., Линч Х.Т. (2010). «Современные гипотезы о том, как микросателлитная нестабильность приводит к увеличению выживаемости пациентов с синдромом Линча» . Клин. Дев. Иммунол . 2010 : 1–13. дои : 10.1155/2010/170432 . ПМК 2901607 . ПМИД 20631828 .
- ^ Кункель Т.А., Эри Д.А. (2005). «Устранение несоответствия ДНК» . Анну. Преподобный Биохим . 74 : 681–710. doi : 10.1146/annurev.biochem.74.082803.133243 . ПМИД 15952900 .
- ^ Кастринос Ф., Сингал С. (2011). «Наследственные синдромы колоректального рака» . Раковый журнал . 17 (6): 405–15. дои : 10.1097/PPO.0b013e318237e408 . ПМК 3240819 . ПМИД 22157284 .
- ^ Нойманн Х.П., Павлу С., Печковска М., Бауш Б., Маквинни С.Р., Муресан М., Бухта М., Франке Г., Клиш Дж., Блей Т.А., Хогерле С., Бедекер CC, Опочер Г., Шиппер Дж., Янушевич А., Энг С. (2004) . «Отличные клинические особенности синдромов параганглиомы, связанных с мутациями генов SDHB и SDHD» . ДЖАМА . 292 (8): 943–51. дои : 10.1001/jama.292.8.943 . ПМИД 15328326 .
- ^ Малкин Д. (апрель 2011 г.). «Синдром Ли-фраумени» . Гены рака . 2 (4): 475–84. дои : 10.1177/1947601911413466 . ПМЦ 3135649 . ПМИД 21779515 .
- ^ Бакри, Д. (2013). P53 в клинике: Зародышевые мутации TP53: генетика синдрома Ли-Фраумени . Нью-Йорк: Спрингер. стр. 167–188. ISBN 978-1-4614-3676-8 .
- ^ Берч Дж. М. (июль 1994 г.). «Семейные раковые синдромы и кластеры». Британский медицинский бюллетень . 50 (3): 624–39. doi : 10.1093/oxfordjournals.bmb.a072913 . ПМИД 7987644 .
- ^ Кенель С., Малкин Д. (август 1997 г.). «Генетическая предрасположенность к раку и семейным раковым синдромам». Педиатр. Клин. Северный Ам . 44 (4): 791–808. дои : 10.1016/s0031-3955(05)70530-7 . ПМИД 9286285 .
- ^ https://doi.org/10.1158/1078-0432.CCR-17-0408.
- ^ Сэмпсон-младший, Джонс Н. (2009). «МУТИХ-ассоциированный полипоз». Best Pract Res Clin Гастроэнтерол . 23 (2): 209–18. дои : 10.1016/j.bpg.2009.03.006 . ПМИД 19414147 . S2CID 24117301 .
- ^ Манфреди М., Вескови П., Бонанини М., Портер С. (март 2004 г.). «Синдром невоидной базальноклеточной карциномы: обзор литературы». Международный журнал челюстно-лицевой хирургии . 33 (2): 117–24. дои : 10.1054/ijom.2003.0435 . ПМИД 15050066 .
- ^ Ло Музио Л (2008). «Синдром невоидной базальноклеточной карциномы (синдром Горлина)» . Сиротский журнал редких заболеваний . 3:32 . дои : 10.1186/1750-1172-3-32 . ПМК 2607262 . ПМИД 19032739 .
- ^ Ричард, С; Гарди, Б; Куве, С; Гад, С. (30 мая 2012 г.). «Фон Хиппель-Линдау: Как редкое заболевание проливает свет на биологию рака». Семинары по биологии рака . 23 (1): 26–37. doi : 10.1016/j.semcancer.2012.05.005 . ПМИД 22659535 .
- ^ Генри, Тодд; Кэмпелл, Джеймс; Хоули, Артур (1969). Клинический диагноз Тодда-Сэнфорда с помощью лабораторных методов под редакцией Исраэля Дэвидсона [и] Джона Бернарда Генри (14-е изд.). Филадельфия: Сондерс. п. 555. ИСБН 978-0-7216-2921-6 .
- ^ Вонг WT, n E, Agró Coleman HR и др. (февраль 2007 г.). «Корреляция генотипа-фенотипа при болезни фон Гиппеля-Линдау с ангиоматозом сетчатки» . Архив офтальмологии . 125 (2): 239–45. дои : 10.1001/archopht.125.2.239 . ПМК 3019103 . ПМИД 17296901 . Архивировано из оригинала 12 декабря 2008 г. Проверено 22 октября 2008 г.
- ^ Перейти обратно: а б с Леманн А.Р., МакГиббон Д., Стефанини М. (2011). «Пигментная ксеродерма» . Сиротский журнал редких заболеваний . 6:70 . дои : 10.1186/1750-1172-6-70 . ПМК 3221642 . ПМИД 22044607 .
- ^ Нидернхофер Л.Дж., Бор В.А., Сандер М., Кремер К.Х. (2011). «Пигментная ксеродерма и другие заболевания преждевременного старения человека и репарация ДНК: молекулы для пациентов» . Мех. Стареющий Дев . 132 (6–7): 340–7. дои : 10.1016/j.mad.2011.06.004 . ПМЦ 3474983 . ПМИД 21708183 .
- ^ Перейти обратно: а б Кеймлинг М., Волчич М., Чернок А., Виланд Б., Дёрк Т., Висмюллер Л. (2011). «Функциональная характеристика связывает индивидуальные мутации пациентов с мутированной атаксией-телеангиэктазией (АТМ) с дисфункцией специфических сигнальных путей двухцепочечного разрыва ДНК» . Журнал ФАСЭБ . 25 (11): 3849–60. дои : 10.1096/fj.11-185546 . ПМИД 21778326 . S2CID 24698475 .
- ^ Томпсон Л.Х., Шильд Д. (2002). «Рекомбинационная репарация ДНК и болезни человека» . Мутат. Рез . 509 (1–2): 49–78. дои : 10.1016/s0027-5107(02)00224-5 . ПМИД 12427531 .
- ^ Нимонкар А.В., Озсой А.З., Геншель Дж., Модрич П., Ковальчиковски С.К. (2008). «Человеческая экзонуклеаза 1 и хеликаза BLM взаимодействуют, разрезая ДНК и инициируя восстановление ДНК» . Учеб. Натл. акад. наук. США . 105 (44): 16906–11. Бибкод : 2008PNAS..10516906N . дои : 10.1073/pnas.0809380105 . ПМЦ 2579351 . ПМИД 18971343 .
- ^ Герман Дж (1969). «Синдром Блума. I. Генетические и клинические наблюдения у первых двадцати семи больных» . Американский журнал генетики человека . 21 (2): 196–227. ПМК 1706430 . ПМИД 5770175 .
- ^ Нагараджу Дж., Скалли Р. (2007). «Учитывая пробел: подземные функции BRCA1 и BRCA2 в остановившихся вилках репликации» . Восстановление ДНК (Амст.) . 6 (7): 1018–31. дои : 10.1016/j.dnarep.2007.02.020 . ПМК 2989184 . ПМИД 17379580 .
- ^ Ланкастер Дж. М., Пауэлл CB, Чен Л. М., Ричардсон Д. Л. (2015). «Заявление Общества гинекологической онкологии об оценке риска наследственной предрасположенности к гинекологическому раку». Гинекол. Онкол . 136 (1): 3–7. дои : 10.1016/j.ygyno.2014.09.009 . ПМИД 25238946 .
- ^ Томпсон Л.Х., Хинц Дж.М. (2009). «Клеточные и молекулярные последствия дефектных белков анемии Фанкони в репликационно-связанной репарации ДНК: понимание механизма» . Мутат. Рез . 668 (1–2): 54–72. дои : 10.1016/j.mrfmmm.2009.02.003 . ПМК 2714807 . ПМИД 19622404 .
- ^ Альтер БП (2003). «Рак при анемии Фанкони, 1927-2001 гг.» . Рак . 97 (2): 425–40. дои : 10.1002/cncr.11046 . ПМИД 12518367 . S2CID 38251423 .
- ^ Мейер Л.А., Броддус Р.Р., Лу К.Х. (2009). «Рак эндометрия и синдром Линча: клинические и патологические аспекты» . Контроль рака . 16 (1): 14–22. дои : 10.1177/107327480901600103 . ПМЦ 3693757 . ПМИД 19078925 .
- ^ Каретерс Дж. М., Стоффель Э. М. (2015). «Синдром Линча и имитаторы синдрома Линча: растущая сложная картина наследственного рака толстой кишки» . Всемирный журнал гастроэнтерологии . 21 (31): 9253–61. дои : 10.3748/wjg.v21.i31.9253 . ПМЦ 4541378 . ПМИД 26309352 .
- ^ Кастан М.Б. (2008). «Реакция на повреждение ДНК: механизмы и роль в заболеваниях человека: лекция на премию Мемориала ГСГ Клоуза 2007 г.» . Мол. Рак Рез . 6 (4): 517–24. дои : 10.1158/1541-7786.MCR-08-0020 . ПМИД 18403632 .
- ^ Викторссон К., Де Петрис Л., Левенсон Р. (2005). «Роль р53 в реакции на лечение рака легких». Биохим. Биофиз. Коммунальный Рес . 331 (3): 868–80. дои : 10.1016/j.bbrc.2005.03.192 . ПМИД 15865943 .
- ^ Теста-младший, Малкин Д., Шиффман Дж.Д. (2013). «Связь молекулярных путей с наследственными синдромами риска рака» . Учебная книга Американского общества клинической онкологии . 33 : 81–90. дои : 10.1200/EdBook_AM.2013.33.81 . ПМК 5889618 . ПМИД 23714463 .
- ^ Рапп А, Грейлих КО (2004). «После индукции двухцепочечного разрыва УФ-А гомологичная рекомбинация и негомологичное соединение концов взаимодействуют в одном и том же DSB, если обе системы доступны» . Журнал клеточной науки . 117 (Часть 21): 4935–45. дои : 10.1242/jcs.01355 . ПМИД 15367581 .
- ^ Барткова Дж, Томмиска Дж, Оплюстилова Л, Аалтонен К, Тамминен А, Хейккинен Т, Мистрик М, Айттомяки К, Бломквист С, Хейккиля П, Лукас Дж, Неванлинна Х, Бартек Дж (2008). «Аберрации сенсорного комплекса повреждения ДНК MRE11-RAD50-NBS1 при раке молочной железы человека: MRE11 как кандидатный ген, предрасполагающий к семейному раку» . Мол Онкол . 2 (4): 296–316. дои : 10.1016/j.molonc.2008.09.007 . ПМЦ 5527773 . ПМИД 19383352 .
- ^ Маркканен Э., Дорн Дж., Хюбшер У (2013). «ДНК-гликозилаза MUTYH: обоснование удаления неповрежденных оснований из ДНК» . Фронт Генет . 4 : 18. doi : 10.3389/fgene.2013.00018 . ПМЦ 3584444 . ПМИД 23450852 .
- ^ Патель С.Г., Анен DJ (2012). «Семейные синдромы рака толстой кишки: обновленная информация в быстро развивающейся области» . Карр Гастроэнтерол Представитель . 14 (5): 428–38. дои : 10.1007/s11894-012-0280-6 . ПМК 3448005 . ПМИД 22864806 .
- ^ Перейти обратно: а б Хшановска К.Х., Грегорек Х., Дембовска-Багиньска Б., Калина М.А., Дигвид М. (2012). «Синдром разрушения Неймегена (NBS)» . Сиротский журнал редких заболеваний . 7:13 . дои : 10.1186/1750-1172-7-13 . ПМЦ 3314554 . ПМИД 22373003 .
- ^ Крокан Х.Э., Бьорос М. (2013). «Базовый эксцизионный ремонт» . Колд Спринг Харб Перспектива Биол . 5 (4): а012583. doi : 10.1101/cshperspect.a012583 . ПМЦ 3683898 . ПМИД 23545420 .
- ^ Койпер Р.П., Хугербрюгге Н. (2015). «NTHL1 определяет новый раковый синдром» . Онкотаргет . 6 (33): 34069–70. дои : 10.18632/oncotarget.5864 . ПМЦ 4741436 . ПМИД 26431160 .
- ^ Сингх Д.К., Ан Б., Бор В.А. (2009). «Роль RECQ-хеликаз в рекомбинационной репарации ДНК, стабильности генома и старении» . Биогеронтология . 10 (3): 235–52. дои : 10.1007/s10522-008-9205-z . ПМЦ 2713741 . ПМИД 19083132 .
- ^ Анбари К.К., Иерарди-Курто Л.А., Силбер Дж.С., Асада Н., Спиннер Н., Закай Э.Х., Беласко Дж., Морриссетт Дж.Д., Дорманс Дж.П. (2000). «Две первичные остеосаркомы у пациента с синдромом Ротмунда-Томсона». Клин. Ортоп. Отн. Рез . 378 (378): 213–23. дои : 10.1097/00003086-200009000-00032 . ПМИД 10986997 . S2CID 36781050 .
- ^ Бор В.А. (2005). «Недостаточная репарация ДНК при прогероидном заболевании человека, синдроме Вернера» . Мутат. Рез . 577 (1–2): 252–9. дои : 10.1016/j.mrfmmm.2005.03.021 . ПМИД 15916783 .
- ^ Моннат Р.Ж. (2010). «Хеликазы RECQ человека: роль в метаболизме ДНК, мутагенезе и биологии рака» . Семин. Рак Биол . 20 (5): 329–39. дои : 10.1016/j.semcancer.2010.10.002 . ПМК 3040982 . ПМИД 20934517 .
- ^ Перейти обратно: а б Менк, Манфорд В. (2014). «Болезни восстановления ДНК: что они говорят нам о раке и старении?» . Жене. Мол. Биол . 37 (1 приложение): 220–33. дои : 10.1590/s1415-47572014000200008 . ПМЦ 3983582 . ПМИД 24764756 .
- ^ Перейти обратно: а б О К.С., Имото К., Эммерт С., Тамура Д., ДиДжованна Дж.Дж., Кремер К.Х. (2011). «Белки эксцизионной репарации нуклеотидов быстро накапливаются, но не сохраняются в клетках XP-E человека (мутант DDB2)» . Фотохим. Фотобиол . 87 (3): 729–33. дои : 10.1111/j.1751-1097.2011.00909.x . ПМК 3082610 . ПМИД 21388382 .
- ^ Перейти обратно: а б Оплеталова К., Бурильон А., Ян В., Пувель С., Армье Дж., Деспрас Е., Людовик М., Матеус С., Роберт С., Каннуш П., Суфир Н., Сарасин А. (2014). «Корреляция фенотипа/генотипа в когорте из 23 пациентов с пигментным вариантом ксеродермии выявила 12 новых болезнетворных мутаций POLH» . Хм. Мутат . 35 (1): 117–28. дои : 10.1002/humu.22462 . ПМИД 24130121 . S2CID 2854418 .
- ^ Перейти обратно: а б Фулкс, Уильям Д.; Кнопперс, Барта Мария; Тернбулл, Клэр (январь 2016 г.). «Популяционное генетическое тестирование на предрасположенность к раку: мутации-основатели геномов». Обзоры природы. Клиническая онкология . 13 (1): 41–54. дои : 10.1038/nrclinonc.2015.173 . ISSN 1759-4782 . ПМИД 26483301 . S2CID 24456816 .
- ^ Справочник, Дом генетики. «Что такое генетическое тестирование?» . Домашний справочник по генетике . Проверено 20 февраля 2018 г.
- ^ Майерс, Мелани Ф.; Бернхардт, Барбара А. (июнь 2012 г.). «Генетическое тестирование непосредственно потребителю: введение в специальный выпуск» . Журнал генетического консультирования . 21 (3): 357–360. дои : 10.1007/s10897-012-9500-3 . ISSN 1573-3599 . ПМИД 22441809 . S2CID 18281938 .
- ^ Справочник, Дом генетики. «Какова стоимость генетического тестирования и сколько времени занимает получение результатов?» . Домашний справочник по генетике . Проверено 20 февраля 2018 г.
- ^ Робсон, Марк Э.; Брэдбери, Анджела Р.; Арун, Бану; Домчек, Сьюзен М.; Форд, Джеймс М.; Хэмпель, Хизер Л.; Липкин, Стивен М.; Сингал, Сапна; Воллинз, Дана С. (1 ноября 2015 г.). «Обновление заявления о политике Американского общества клинической онкологии: генетическое и геномное тестирование на восприимчивость к раку». Журнал клинической онкологии . 33 (31): 3660–3667. дои : 10.1200/JCO.2015.63.0996 . ISSN 1527-7755 . ПМИД 26324357 .
- ^ «Генетическое тестирование на риск рака» . Исследования рака Великобритании . 02.06.2015 . Проверено 20 февраля 2018 г.
- ^ Шраг, Д.; Кунц, К.М.; Гарбер, Дж. Э.; Уикс, Джей Си (15 мая 1997 г.). «Анализ решений - влияние профилактической мастэктомии и овариэктомии на продолжительность жизни женщин с мутациями BRCA1 или BRCA2» . Медицинский журнал Новой Англии . 336 (20): 1465–1471. дои : 10.1056/NEJM199705153362022 . ISSN 0028-4793 . ПМИД 9148160 .
- ^ Ньютон, К.; Грин, К.; Лаллоо, Ф.; Эванс, генеральный директор; Хилл, Дж. (январь 2015 г.). «Приверженность и результаты колоноскопического скрининга у пациентов с синдромом Линча». Колоректальное заболевание . 17 (1): 38–46. дои : 10.1111/codi.12778 . ISSN 1463-1318 . ПМИД 25213040 . S2CID 205027427 .
- ^ Казак, Матвей; Гаффари, Кэмерон; Уотсон, Патрис; Снайдер, Кэрри; Линч, Генри (апрель 2014 г.). «Употребление аспирина связано с более низким риском рака простаты у мужчин-носителей мутаций BRCA» . Журнал генетического консультирования . 23 (2): 187–191. дои : 10.1007/s10897-013-9629-8 . ISSN 1573-3599 . ПМИД 23881471 . S2CID 15371573 .
- ^ Торат, Мангеш А.; Кьюзик, Джек (декабрь 2013 г.). «Роль аспирина в профилактике рака» . Текущие отчеты по онкологии . 15 (6): 533–540. дои : 10.1007/s11912-013-0351-3 . ISSN 1534-6269 . ПМИД 24114189 . S2CID 40187047 .
- ^ Справочник, Дом генетики. «Синдром Вернера» . Домашний справочник по генетике . Проверено 20 февраля 2018 г.
- ^ «Генетический риск, раса и этническая принадлежность | Журнал Cancer Fighters Thrive Magazine» . CancerCenter.com . Архивировано из оригинала 21 февраля 2018 г. Проверено 20 февраля 2018 г.