Семейный аденоматозный полипоз
| Семейный аденоматозный полипоз | |
|---|---|
| Другие имена | ФАП |
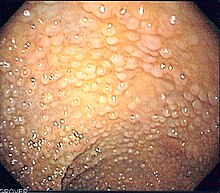 | |
| Эндоскопическая картина сигмовидной кишки больного семейным аденоматозным полипозом | |
| Специальность | Гастроэнтерология , Онкология |
| Осложнения | Колоректальный рак |
| Обычное начало | <35 лет |
| Продолжительность | Пожизненный |
| Типы | Классический или ослабленный |
| Причины | Мутация гена APC |
| Метод диагностики | Колоноскопия Генетическое тестирование |
| Дифференциальный диагноз | Синдром Линча , MUTYH-ассоциированный полипоз |
| Уход | Колоноскопия Полипэктомия Верхняя эндоскопия Колэктомия |
| Частота | 1 из 10 000 - 15 000 |
Семейный аденоматозный полипоз ( САП ) — аутосомно-доминантное наследственное заболевание, при котором многочисленные аденоматозные полипы формируются преимущественно в эпителии толстого кишечника . Хотя эти полипы изначально доброкачественные , злокачественная трансформация в рак толстой кишки при отсутствии лечения происходит . Известно, что существуют три варианта: FAP и ослабленный FAP (первоначально называемый синдромом наследственной плоской аденомы) . [1] ) вызваны APC дефектами гена на хромосоме 5, тогда как аутосомно-рецессивный FAP (или MUTYH-ассоциированный полипоз ) вызван дефектами гена MUTYH на хромосоме 1. Из этих трех сам FAP является наиболее тяжелым и наиболее распространенным; хотя во всех трех случаях возникающие полипы и рак толстой кишки первоначально ограничиваются стенкой толстой кишки. Обнаружение и удаление до появления метастазов за пределами толстой кишки может значительно снизить, а во многих случаях и полностью исключить распространение рака.
Основной причиной САП считается генетическая мутация в организме — изменение генов-супрессоров опухолей , которые предотвращают развитие опухолей. Это изменение позволяет многочисленным клеткам кишечной стенки развиться в потенциально раковые полипы, когда они обычно достигают конца своей жизни; неизбежно один или несколько из них в конечном итоге прогрессируют и приводят к раку (риск 7% к 21 году, рост до 87% к 45 годам и 93% к 50 годам). Эти генные изменения не вызывают рак, а, скорее, снижают способность организма предотвращать превращение клеток в раковые. Даже при изменении гена может пройти некоторое время, прежде чем действительно разовьется клетка, которая в результате станет раковой, и в некоторых случаях ген все еще может частично контролировать опухоли, поэтому рак из-за FAP развивается в течение многих лет и почти всегда заболевание, начинающееся во взрослом возрасте.
Вторая форма FAP, известная как ослабленный семейный аденоматозный полипоз, имеет функциональный ген APC , но слегка нарушенный. Поэтому он в некоторой степени может работать как обычно. Ослабленный FAP по-прежнему представляет высокий 70%-ный риск развития рака в течение жизни (по оценкам), но обычно проявляется гораздо меньшим количеством полипов (обычно 30), а не сотнями или тысячами, обычно встречающимися при FAP. [2] и возникает в возрасте, когда ФАП обычно уже не считается вероятным — обычно между 40 и 70 годами (в среднем 55 лет). [3] ), а не более обычные 30 и выше. Поскольку полипов гораздо меньше, варианты лечения могут быть разными. [2]
Третий вариант, аутосомно-рецессивный семейный аденоматозный полипоз или MUTYH-ассоциированный полипоз , также является более легким и, как следует из названия, требует, чтобы оба родителя были «носителями» для проявления заболевания.
В некоторых случаях САП может проявляться в толстой кишке выше обычного (например, восходящая ободочная кишка , [ нужна ссылка ] или проксимальнее селезеночного изгиба , или в желудке или двенадцатиперстной кишке. [1] ), где они не проявляют никаких симптомов до тех пор, пока рак не появится и не достигнет значительной стадии. Мутации APC связаны с некоторыми другими видами рака, такими как рак щитовидной железы . Поскольку мутация, вызывающая ФАП, является аутосомно-доминантной, она может быть унаследована непосредственно от любого родителя ребенку. Существует генетический анализ крови на ген APC , который может определить, присутствует ли он, и, следовательно, может предсказать возможность САП. Лицам из группы риска (из-за семейных связей или генетического тестирования) обычно предлагается плановый мониторинг кишечного тракта каждые 1–3 года на протяжении всей жизни, начиная с полового созревания при САП и в раннем взрослом возрасте при ослабленных формах. Операция по резекции толстой кишки рекомендуется при обнаружении многочисленных полипов толстой кишки из-за высокого риска ранней смерти от рака толстой кишки. Существуют международные регистры полипоза, которые отслеживают известные случаи дефектов генов FAP или APC для исследовательских и клинических целей. Мутация APC также часто встречается в случаях колоректальной карциномы, что подчеркивает ее важность при этой форме рака.
Признаки и симптомы
[ редактировать ]Начиная с раннего подросткового возраста, у пациентов с этим заболеванием постепенно (и большую часть времени бессимптомно) развиваются сотни и тысячи колоректальных полипов (а иногда и полипов в других местах ) — небольших аномалий на поверхности кишечного тракта , особенно в толстой кишке , включая толстую или толстую кишку. прямая кишка . Они могут кровоточить, что приводит к появлению крови в стуле. Если крови не видно, у больного все равно возможно развитие анемии из-за постепенно развивающегося дефицита железа. Если развивается злокачественное новообразование, это может проявляться потерей веса , изменением режима работы кишечника или даже метастазами в печень или где-либо еще. САП также может развиваться «тихо» у некоторых людей, проявляя мало признаков или вообще не проявляя их, пока не перерастет в запущенный колоректальный рак . [ нужна ссылка ]
Поскольку семейный полипоз развивается очень постепенно в течение многих лет, а также может проявляться в «ослабленной» форме еще более постепенно, полипы, возникающие в результате САП, могут привести к развитию рака в любой момент, от подросткового возраста до старости. [ нужна ссылка ]
В зависимости от характера дефекта гена APC, а также от того, является ли он полной или ослабленной формой, семейный полипоз может проявляться в виде полипов в толстой кишке или двенадцатиперстной кишке или в любой их комбинации. Следовательно, отсутствие полипов, например, в прямой кишке, само по себе может быть недостаточным для подтверждения отсутствия полипов. Может возникнуть необходимость рассмотреть и визуально осмотреть другие возможные отделы кишечного тракта. В этом отношении колоноскопия предпочтительнее ректороманоскопии , поскольку она обеспечивает лучшее наблюдение за частым правосторонним расположением полипов. [1]

Генетическая детерминанта семейного полипоза может также предрасполагать носителей к другим злокачественным новообразованиям, например двенадцатиперстной кишки и желудка (особенно ампулярной аденокарциноме). Другими признаками, которые могут указывать на САП, являются развитие фибромы Гарднера и десмоидных опухолей (доброкачественные опухоли кожи, которые могут проявляться раньше других признаков САП), [4] пигментные поражения сетчатки ( «ВГРПЭ — врожденная гипертрофия пигментного эпителия сетчатки»), кисты челюстей, сальные кисты и остеомы (доброкачественные опухоли костей). Сочетание полипоза, остеом, фибром и кист сальных желез называется синдромом Гарднера (с аномальным рубцеванием или без него). [5]
Генетика
[ редактировать ]Семейный аденоматозный полипоз может иметь разные закономерности наследования и разные генетические причины. Когда это состояние возникает в результате мутации гена , APC оно наследуется по аутосомно-доминантному типу, что означает, что одной копии измененного гена достаточно, чтобы вызвать заболевание. Частота злокачественных новообразований в этих случаях приближается к 100%. В большинстве случаев у пострадавшего есть один из родителей с этим заболеванием. [ нужна ссылка ]
Варианты мутации гена APC
[ редактировать ]APC ответственный — это ген-супрессор опухоли, за выработку аденоматозного полипоза coli (APC), большого многофункционального белка, подавляющего опухоль, который действует как «привратник», предотвращая развитие опухолей. (APC регулирует β-катенин , белок, который играет решающую роль в клеточной коммуникации, передаче сигналов, росте и контролируемом разрушении, но который, если оставить его без контроля, также приводит к многочисленным видам рака). [1] ). Изъян в гене APC означает, что APC не так эффективен, как должен быть, и вполне вероятно, что со временем некоторые клетки, которые должны были контролироваться APC, перестанут контролироваться и вместо этого продолжат развиваться и станут раковыми. При знакомом полипозе они обычно проявляются в виде полипов — небольших аномалий на поверхности кишечного тракта . [ нужна ссылка ]
Хотя полипы по своей природе доброкачественные, первый шаг гипотезы двух ударов уже произошел: унаследованная мутация APC. Часто оставшийся «нормальный» аллель мутирует или удаляется, что ускоряет образование полипов. Дальнейшие мутации (например, в p53 или kRAS ) в APC-мутированных клетках с гораздо большей вероятностью приведут к раку, чем в немутированных эпителиальных клетках. [ нужна ссылка ]
Нормальная функция продукта гена APC все еще исследуется; он присутствует как в ядре клетки , так и в мембране. Канонической опухолесупрессорной функцией APC является подавление β-катенина, но другие опухолесупрессорные функции APC могут быть связаны с адгезией клеток и организацией цитоскелета .
Мутация APC также часто встречается в случаях колоректальной карциномы, что подчеркивает ее важность при этой форме рака.
Варианты мутации гена MUTYH
[ редактировать ]MUTYH кодирует репарации ДНК фермент MYH гликозилазу . Во время нормальной клеточной деятельности гуанин иногда изменяется под действием кислорода , что приводит к его соединению с аденином вместо цитозина . Гликозилаза MYH исправляет эти ошибки путем вырезания оснований , так что мутации не накапливаются в ДНК и не приводят к образованию опухоли. Когда гликозилаза MYH не функционирует правильно, могут накапливаться ошибки ДНК, инициирующие онкогенез с клинической картиной, аналогичной таковой у пациентов с мутациями APC. [ нужна ссылка ]
Мутации в гене MUTYH наследуются по аутосомно-рецессивному типу, что означает, что для того, чтобы у человека появилось заболевание, должны быть изменены две копии гена. Чаще всего родители ребенка с аутосомно-рецессивным заболеванием не страдают, но являются носителями одной копии измененного гена.
Модели животных
[ редактировать ]« АПК Мин Мышиная модель была описана в 1990 году и несет аллель Apc со стоп-кодоном в положении 850. Гетерозиготность по этой мутации приводит к полностью проникающему фенотипу на большинстве генетических фонов, при этом у мышей на чувствительном фоне развивается более 100 опухолей в кишечном тракте. Количество и расположение опухолей кишечника модифицируются несвязанными генами. С тех пор появилось множество других моделей, включая модель аттенуированного FAP (модель 1638N) и несколько условных мутантов , которые позволяют тканеспецифическую или временную абляцию функции гена. дополнительную информацию см. в моделях колоректального и кишечного рака на мышах . [ нужна ссылка ]
В 2007 году на крысиной модели «ApcPirc» был выделен стоп-кодон в положении 1137. [6] В отличие от мышиных моделей, где >90% опухолей образуются в тонком кишечнике, у крыс Пирца опухоли преимущественно (>60%) образуются в толстом кишечнике, что аналогично клинической картине у человека.
Диагностика
[ редактировать ]
Постановка диагноза САП до развития рака толстой кишки важна не только для человека, но и для других членов семьи, которые могут быть затронуты. Существует два метода диагностики: [ нужна ссылка ]
- Колоноскопия является обычным диагностическим тестом выбора, поскольку она лучше, чем ректороманоскопия, способствует распространенному правостороннему расположению полипов, если мутация ослаблена FAP. [1] и может подтвердить или разрешить (а) фактическую клиническую картину и любое изменение состояния человека, находящегося в группе риска, (б) количественную оценку полипов по всей толстой кишке, (в) гистологический диагноз (обнаружение типа клеток/рака) и (г) при наличии полипов это может указывать на целесообразность амбулаторного иссечения (удаления) или рекомендуется хирургическое вмешательство. Бариевая клизма и виртуальная колоноскопия (разновидность медицинской визуализации) также могут использоваться для подтверждения диагноза САП.
- Генетическое тестирование позволяет поставить окончательный диагноз в 95% случаев; генетическое консультирование обычно необходимо в семьях, где был диагностирован ФАП. Тестирование также может помочь в диагностике пограничных случаев в семьях, в которых иначе известны p34.3 и p32.1 (1p34.3–p32.1). Тестирование может только показать, восприимчив ли человек к FAP, или исключить его (т. е. унаследовал ли он дефектный ген APC). Он не может определить фактическое состояние пациента; это можно обнаружить только при прямом физическом осмотре.
NCBI заявляет, что врачи должны убедиться, что они понимают «риски, преимущества и ограничения» любого генетического теста, проведенного с 1997 года, «почти для трети людей, прошедших обследование на ФАП, врачи неверно истолковали результаты теста». [7]
После установления диагноза САП тщательное колоноскопическое наблюдение с полипэктомией необходимо .
Пренатальное тестирование возможно, если у пораженного члена семьи выявлена мутация, вызывающая заболевание; однако пренатальное тестирование на расстройства, возникающие, как правило, у взрослых, проводится редко и требует тщательного генетического консультирования .
УЗИ брюшной полости и анализы крови, оценивающие функцию печени, часто проводятся, чтобы исключить метастазы в печень.
Управление
[ редактировать ]
Из-за того, как развивается семейный полипоз, можно иметь генетическое заболевание и, следовательно, подвергаться риску, но пока не иметь полипов или проблем. Таким образом, у человека может быть диагностирован «риск» САП, и ему может потребоваться регулярное наблюдение, но он (пока) фактически не имеет ФАП (т. е. несет дефектный ген, но, похоже, пока не имеет каких-либо реальных медицинских проблем в результате этого). ). Клиническое ведение может охватывать несколько областей: [ нужна ссылка ]
- Выявление тех людей, которые могут подвергаться риску САП: обычно на основании семейного медицинского анамнеза или генетического тестирования.
- Диагностика (подтверждение наличия у них САП) — это можно сделать либо с помощью генетического тестирования, которое является окончательным, либо путем визуальной проверки самого кишечного тракта.
- Важно отметить, что визуальный осмотр или мониторинг не могут «очистить» человека от риска. Он может только сказать, каково их состояние на данный момент. Если в какой-то момент жизни у человека появляются многочисленные полипы, это может указывать на диагноз САП. (Отсутствие полипов не «очищает» человека, поскольку полипы могут развиться в более позднем возрасте; также несколько полипов с течением времени не так уж редки у людей без ФАП. Однако значительное количество или изобилие полипов обычно указывают на диагностика САП и гистопатология , чтобы определить, являются ли какие-либо полипы раковыми.)
- Программы скрининга/мониторинга включают визуальное обследование кишечного тракта для проверки его здорового состояния. Оно проводится в плановом порядке каждые несколько лет, когда есть повод для беспокойства, когда либо (а) генетический тест подтвердил риск, либо (б) генетический тест не был проведен по какой-либо причине, поэтому фактический риск неизвестен. Скрининг и мониторинг позволяют обнаружить полипоз визуально до того, как он станет опасным для жизни.
- Лечение, обычно хирургическое, применяется, если полипоз привел к большому количеству полипов, значительному риску рака или реальному раку.
Семейная история
[ редактировать ]NCBI заявляет, что «хотя у большинства людей с диагнозом полипоза, связанного с APC, есть пораженный родитель, семейный анамнез может оказаться отрицательным из-за неспособности распознать заболевание у членов семьи, ранней смерти родителя до появления симптомов, или позднее начало заболевания у пострадавшего родителя». [2] Кроме того, около 20% случаев являются мутациями de novo , а из лиц с явной мутацией APC de novo (т.е. при отсутствии известной семейной истории) 20% имеют соматический мозаицизм . [8] Также известно, что существуют бессимптомные люди (и, следовательно, бессимптомные члены семьи). [2]
Мониторинг
[ редактировать ]Мониторинг включает проведение амбулаторной колоноскопии верхних отделов желудка , а иногда и эзофагогастродуоденоскопии (ЭГДС) для выявления предраковых заболеваний желудка или двенадцатиперстной кишки. [1] ), обычно один раз в 1–3 года, и/или генетический анализ крови для окончательного подтверждения или опровержения восприимчивости. Небольшое количество полипов часто можно иссечь (удалить) во время процедуры, если они обнаружены, но если есть более серьезные признаки или количества, стационарное хирургическое вмешательство . может потребоваться [ нужна ссылка ]
NCBI заявляет, что, когда у человека выявляется наличие САП или мутаций, приводящих к САП: «Уместно оценить родителей больного человека (а) с помощью молекулярно-генетического тестирования АПК, если мутация, вызывающая заболевание, известна в пробанд [человек, у которого впервые выявлено это заболевание] или (b) для клинических проявлений состояний полипоза, связанных с АПК». [2]
Уход
[ редактировать ]Лечение САП зависит от генотипа . У большинства людей с мутацией APC рак толстой кишки развивается к 40 годам, хотя менее распространенная ослабленная версия обычно проявляется в более позднем возрасте (40–70 лет). Соответственно, во многих случаях профилактическое хирургическое вмешательство может быть рекомендовано в возрасте до 25 лет или при обнаружении заболевания при активном наблюдении. Существует несколько хирургических вариантов, которые включают удаление толстой кишки или прямой и толстой кишки.
- Поражение прямой кишки: прямая кишка и часть или вся толстая кишка удаляются. Пациенту может потребоваться илеостома (постоянная стома , при которой стул попадает в мешок на животе) или реконструкция илео-анального мешка . Решение об удалении прямой кишки зависит от количества полипов в прямой кишке, а также от семейного анамнеза. Если в прямой кишке мало полипов, толстую кишку частично или полностью удаляют и вместо этого можно напрямую соединить тонкую кишку (подвздошную кишку) ( илеоректальный анастомоз ).
- Прямая кишка не вовлекается: часть толстой кишки с полипами может быть удалена, а концы «воссоединены» ( частичная колэктомия ) — операция, требующая значительного времени заживления, но сохраняющая качество жизни в значительной степени неизмененным.
Профилактическая колэктомия показана при наличии более ста полипов, при наличии сильно диспластических полипов или при наличии множественных полипов размером более 1 см.
Лечение двух более легких форм САП может существенно отличаться от более обычного варианта, поскольку количество полипов гораздо меньше, что дает больше возможностей.
Исследуются различные лекарства для замедления злокачественного перерождения полипов, в первую очередь нестероидные противовоспалительные препараты (НПВП). Было показано, что НПВП значительно уменьшают количество полипов, но обычно не меняют тактику лечения, поскольку полипов все еще слишком много, чтобы их можно было отслеживать и лечить эндоскопически. Препарат эфлорнитин , ингибитор орнитиндекарбоксилазы, обычно используемый для лечения трипаносомоза , исследуется в качестве потенциального профилактического препарата в сочетании с НПВП целекоксибом для лечения САП. Еще один исследуемый агент — сулиндак , также используемый в сочетании с НПВП. [9] [10] [11]
Прогноз
[ редактировать ]До достижения поздних стадий колоректального рака полипы ограничиваются внутренней стенкой и толщиной кишечного тракта, не метастазируют и не «распространяются». Таким образом, при условии обнаружения и контроля САП либо на предраковой стадии, либо когда раковые полипы все еще находятся внутри кишечного тракта, хирургическое вмешательство имеет очень высокий уровень успеха в предотвращении или удалении рака без рецидива, поскольку места, вызывающие рак, удаляются физически полностью хирургическим путем. [ нужна ссылка ]
После операции, если была выполнена частичная колэктомия , необходимо колоноскопическое наблюдение за оставшейся толстой кишкой, поскольку у человека все еще существует риск развития рака толстой кишки. Однако, если бы это произошло, это был бы новый инцидент, вызванный новым развитием полипов в неудаленной части толстой кишки после операции, а не возвращением или метастазированием любого рака, удаленного первоначальной операцией.
Десмоидные опухоли с их инфильтративным характером и потенциальной близостью к жизненно важным структурам являются второй по значимости причиной смерти. [12]
Эпидемиология
[ редактировать ]Частота мутации составляет от 1 на 10 000 до 1 на 15 000 рождений. К возрасту 35 лет у 95% лиц с САП (>100 аденом) наблюдаются полипы. Без колэктомии рак толстой кишки практически неизбежен. Средний возраст рака толстой кишки у нелеченых лиц составляет 39 лет (диапазон 34–43 года). [13]
Ослабленный FAP возникает, когда APC неисправен, но все еще функционирует. В результате он сохраняет часть своей способности подавлять полипы. Таким образом, ослабленный FAP проявляется как колоректальный рак необычно поздно (возраст 40–70 лет, средний показатель = 55 лет). [3] ), и обычно с небольшим количеством или, по крайней мере, гораздо меньшим количеством полипов (обычно 30 [2] ), чем более обычная версия FAP, в возрасте, когда FAP больше не считается большой вероятностью или риском согласно обычной эпидемиологии FAP.
Сравнение вариантов ФАП
[ редактировать ]В этой таблице сравниваются различные подтипы ФАП: [2] [1]
| Элемент | ФАП | Ослабленный ФАП | МУТЫХ Ассоциированный ФАП |
| Ген | БТР | БТР | МУТЫХ |
| Типичное проявление полипа | Сотни/тысячи | Менее 100 (0–470, тип. 30), иногда плоская, а не полипоидная морфология и более проксимальнее селезеночного изгиба. В исследовании с участием 120 человек у 37% (N=44) было <10 полипов; У троих из этих 44 был колоректальный рак. [14] Также наблюдаются полипы фундального отдела желудка и аденомы двенадцатиперстной кишки. Таким образом, полипы и рак могут проявляться в верхней части толстой кишки или верхних отделах желудочно-кишечного тракта, а не в обычных местах. | ? |
| Типичные основные диагностические критерии | (а) 100+ полипов и возраст до 40 лет, ИЛИ (б) полипы и САП у родственника | Еще не решено. (a) отсутствие в семейном анамнезе более 100 полипов до 30 лет ПЛЮС ОДИН ИЗ 10–99 полипов / более 100 полипов в возрасте от 35 до 40 лет / колоректальный рак до 60 лет и родственники с множественными аденоматозными полипами, ИЛИ (b) семейный анамнез От 10 до 99 аденом диагностируются после 30 лет. | ? |
| Возраст, в котором проявляются полипы | 7–36 (тип. 16), затем быстро увеличивается | ? | ? |
| Риск колоректального рака ( пенетрантность ) и возраст при отсутствии лечения | «неизбежно... практически 100%»: 7% к 21 году, 87% к 45 годам, 93% к 50 годам. Типичный возраст: 34–43 года (в среднем 39). | «Ниже… менее известно… примерно 70% к 80 годам». Совариа заявляет, что по состоянию на 1998 год «средний возраст постановки диагноза КРР составляет ~ 58 лет». | ? |
| Вариативность | Меж- и внутрисемейная фенотипическая изменчивость распространена. | См. ФАП | ? |
| Возможные проявления, не связанные с толстой кишкой | «полипы дна желудка и двенадцатиперстной кишки, остеомы, зубные аномалии, врожденная гипертрофия пигментного эпителия сетчатки (CHRPE), опухоли мягких тканей, десмоидные опухоли и ассоциированные раковые заболевания» | Что касается ФАП, но «CHRPE и десмоидные опухоли встречаются редко», а также добавляется рак щитовидной железы. | ? |
| Другие пожизненные риски | «Рак тонкой кишки [двенадцатиперстной кишки или периампулы] 4–12% [дистальнее двенадцатиперстной кишки] Редко; Аденокарцинома поджелудочной железы ~ 1%; Папиллярный рак щитовидной железы 1–2%; ЦНС [тип. медуллобластома] <1%; Гепатобластома печени 1,6%; Желчь Аденокарцинома протоков Низкая, но повышенная Аденокарцинома желудка <1% в западных культурах». | ? | ? |
| Наследование | «наследуется по аутосомно-доминантному типу. Примерно 75–80% людей с полипозом, связанным с APC, имеют пораженного родителя. Потомство больного человека подвергается 50% риску унаследовать мутацию, вызывающую заболевание» | То же, что ФАП | Разный — рецессивный (требуется, чтобы 2 родителя были носителями). |
| Генетический обзор и генетическое обнаружение | «Полное секвенирование генов всех экзонов APC и границ интрон-экзон является наиболее точным доступным клиническим тестом. Большинство мутаций APC являются бессмысленными или мутациями со сдвигом рамки считывания, которые вызывают преждевременное усечение белка APC. Вероятность обнаружения мутации APC очень высока. зависит от тяжести полипоза толстой кишки и семейного анамнеза. ◦Приблизительно у 20% людей наблюдается очевидная мутация APC de novo. Маркеры, используемые для анализа сцепления состояний, связанных с полипозом APC, высокоинформативны и очень тесно связаны с локус APC, таким образом, их можно использовать с точностью более 98% в более чем 95% семей с полипозом, связанным с APC, тестирование на сцепление невозможно для семей с одним пострадавшим, ситуация, которая часто возникает, когда локус APC; у человека имеется генная мутация de novo и нет пораженного потомства. Если не обнаружено заболевания, вызывающего мутацию APC, следует рассмотреть возможность молекулярно-генетического тестирования MUTYH (см. Дифференциальный диагноз). | «Ожидается, что менее 30% людей с аттенуированными фенотипами будут иметь идентифицируемую мутацию APC» (см. также подробности в разделе FAP). | ? |
| Генотип-Фенотип [Основное состояние] | Наиболее частая мутация APC происходит в кодоне 1309 и приводит к большому количеству полипов в раннем возрасте (~ 20). Сообщалось о обильном полипозе (среднее значение = 5000) с мутациями в кодонах 1250–1464. Большинство частичных и полных делеций APC связаны с 100–2000 аденомами толстой кишки, хотя наблюдалось ослабление FAP. Выборочный типичный возраст начала: между кодоном 168 и 1580 (исключая 1309) = 30 лет, 5' кодона 168 и 3' кодона 1580 = 52 года. | Аттенуированный FAP связан с мутациями (обычно усеченными) в 5'-части гена (кодоны 1–177), экзоне 9 и дистальном 3'-конце гена; интерстициальные делеции хромосомы 5q22, включающие APC; частичные и целые делеции гена; и соматический мозаицизм для мутаций APC, которые обычно связаны с классическим FAP. Совариа утверждает, что ослабленный FAP «вызван мутациями в трех различных областях гена APC — 5' - конце в области, охватывающей экзоны 4 и 5, экзоне 9 и крайнем 3' - конце. Фенотипическая экспрессия в этих трех группах родственников варьируется, но определенно мягче, чем при классическом САП», и что полипы прямой кишки редки при ослабленном САП, но еще не подтверждено, означает ли это также снижение риска рака прямой кишки. | ? |
| Генотип-фенотип [Другие внетолстые состояния] | Выраженные внетолстые проявления часто коррелируют (хотя и не полностью) с более дистальными мутациями APC. Общее исследование САП плюс внекишечных симптомов показало : мутации в кодонах 1395–1493 вызывают значительно более высокий уровень развития десмоидных опухолей, остеом и эпидермоидных кист, чем мутации в кодонах 177–452; мутации в кодонах 1395–1493 вызывают значительно более высокий уровень развития десмоидных опухолей и остеом, чем мутации в кодонах 457–1309; ни у одного человека с мутациями в кодонах 177–452 не развились остеомы или периампулярный рак; только у людей с мутациями в кодонах 457–1309 развивалась гепатобластома и/или опухоли головного мозга. Аденомы двенадцатиперстной кишки : четырехкратное увеличение риска при мутациях между кодонами 976 и 1067. Десмоидные опухоли : мутации 3'-кодона 1399 были связаны с развитием десмоидной опухоли с отношением шансов 4,37; десмоидные опухоли у 20% лиц с мутациями 5' кодона 1444, у 49% лиц с мутациями 3' кодона 1444 и у 61% лиц с мутациями кодонов 1445–1580; несколько семей с тяжелыми десмоидными опухолями имели мутации на крайнем 3'-конце; устойчивая ассоциация десмоидных опухолей с мутациями, дистальными к кодону 1444. CHRPE связан с: мутациями между кодонами 311 и 1444; целые делеции гена APC. Рак щитовидной железы и FAP : у 24 человек большинство выявленных мутаций приходились на 5'-кодон 1220 [Cetta et al. 2000]; У 9 из 12 человек мутации APC были выявлены проксимальнее области кластера мутаций (кодоны 1286–1513). Общий обзор литературы (на август 2006 г.) : выявлено 89 субмикроскопических делеций APC (42 частичных и 47 целых генных делеций). Внетолстые находки наблюдались в 36% случаев, без существенных различий между пациентами с частичной и полной делецией гена. | ? | ? |
| Распространенность | «От 2,29 до 3,2 на 100 000 человек. Полипозные состояния, связанные с АПК, исторически составляли около 0,5% всех случаев колоректального рака; эта цифра снижается по мере того, как все больше членов семьи из группы риска проходят успешное лечение после раннего выявления полипов и профилактической колэктомии». | «Вероятно, не диагностируется, учитывая меньшее количество полипов толстой кишки и меньший риск развития колоректального рака по сравнению с классическим САП» | ? |
| Лечение проявлений | Классический FAP: «Колэктомия рекомендуется после появления аденом; колэктомия может быть отложена в зависимости от размера и количества аденоматозных полипов. Колэктомию обычно рекомендуют, когда развилось более 20 или 30 аденом или множественные аденомы с развитой гистологией» | «Может потребоваться колэктомия, но примерно у трети людей количество полипов толстой кишки настолько ограничено, что достаточно наблюдения с периодической колоноскопической полипэктомией» | ? |
| Деятельность по надзору (мониторингу) после установления риска | «Сигмоидоскопия или колоноскопия каждые 1–2 года, начиная с возраста от 10 до 12 лет; колоноскопия при обнаружении полипов; ежегодная колоноскопия, если колэктомия откладывается более чем на год после появления полипов (возраст от 10 до 20 лет с некоторыми более легкими симптомами, может быть рассмотрена отсрочка колэктомии (ЭГДС) в возрасте до 25 лет или до колэктомии и повторение каждые 1–3 года, в некоторых случаях эндоскопическая ретроградная холангиопанкреатография (ЭРХПГ) для выявления аденом общего желчного протока; визуализация кишечника при обнаружении аденомы двенадцатиперстной кишки или перед колэктомией, повторяется каждые 1–3 года в зависимости от результатов скрининга на гепатобластому (оптимальный интервал неизвестен, в одной статье рекомендуется «не реже одного раза в три месяца»); проявлений, пальпацию щитовидной железы с учетом последующего ультразвукового исследования и тонкоигольной аспирации при наличии узлов щитовидной железы» | «Колоноскопия каждые два-три года, начиная с возраста 18-20 лет; эзофагогастродуоденоскопия (ЭГДС), начиная с 25 лет или до колэктомии и повторяемая каждые 1-3 года; в некоторых случаях может потребоваться эндоскопическая ретроградная холангиопанкреатография (ЭРХПГ). для оценки аденом общего желчного протока; ежегодное физическое обследование с пальпацией щитовидной железы с учетом последующего ультразвукового исследования и тонкоигольной аспирации при наличии узлов щитовидной железы. Колэктомия обычно рекомендуется при наличии более 20 или 30 аденом или множественных аденом. с развитой гистологией». По состоянию на 1998 год Совариа заявляет, что «колоноскопию, в отличие от ректороманоскопии, следует рекомендовать для эндоскопического наблюдения из-за правостороннего расположения колоректальных аденом; эндоскопическое наблюдение UGI оправдано в попытке обнаружить предраковые опухоли желудка или двенадцатиперстной кишки; при [ослабленном САП] может потребоваться тотальная колэктомия с илео-ректальным анастомозом только в том случае, если рекомендуется профилактическая колэктомия» | ? |
| Решение о мониторинге | «Раннее выявление может позволить своевременно вмешаться и улучшить окончательный результат; таким образом, уместно наблюдение за бессимптомными детьми из группы риска на предмет ранних проявлений; генетическое тестирование является более экономически эффективным, чем ректороманоскопия, для определения того, кто в семье поражен; люди с диагнозом АПК Состояния, связанные с полипозом, возникающие в результате наличия пораженного родственника, имеют значительно большую продолжительность жизни, чем те люди, у которых диагноз был диагностирован на основании симптомов. Поскольку мониторинг толстой кишки у лиц с риском классического САП начинается уже в возрасте от десяти до 12 лет, молекулярный генетическое тестирование обычно предлагается детям с риском классического САП в возрасте до десяти лет. Генетическое тестирование при рождении также может быть оправдано, поскольку некоторые родители и педиатры могут рассмотреть возможность проведения скрининга гепатобластомы с младенчества до пятилетнего возраста у пораженного потомства. Никакие доказательства не указывают на это. оптимальный возраст для начала скрининга». | См. ФАП. Также «скрининг толстой кишки для людей с ослабленным FAP начинается в возрасте от 18 до 20 лет; таким образом, молекулярно-генетическое тестирование следует предлагать тем, кто находится в группе риска по ослабленному FAP, примерно в возрасте 18 лет». | ? |
| Наследование и последствия подтвержденного диагноза для других близких родственников | Состояния полипоза, ассоциированного с АПК, наследуются по аутосомно-доминантному типу. Примерно у 20–25% ген изменен в результате генной мутации de novo. Мало или совсем нет доказательств предвзятости по отношению к матери/отцу или эффекта, связанного с преклонным возрастом отца, при de novo мутациях . У братьев и сестер классический 50-процентный риск заражения этим заболеванием, если он унаследован, а не de novo , и «низкий», но немного более высокий риск, чем общий, если de novo , поэтому следует предложить генетическое тестирование. Каждый потомок имеет 50% шанс на наследование. Другие члены семьи подвергаются риску, если их родители имеют ту же мутацию. Зародышевый мозаицизм был зарегистрирован в бессимптомных случаях. Пренатальное тестирование возможно с помощью ДНК , выделенной из плода . | См. ФАП | ? |
Регистры полипоза
[ редактировать ]Из-за генетической природы ФАП во всем мире созданы реестры полипоза. Целью этих реестров является расширение знаний о заразности ФАП, а также документирование, отслеживание и уведомление членов семей заболевших лиц. Одно исследование показало, что использование реестра для уведомления членов семьи (вызовов) значительно снижает смертность по сравнению с пробандами . [15] Реестр полипоза Святого Марка является старейшим в мире, он был основан в 1924 году, и в настоящее время существует множество других реестров полипоза .
См. также
[ редактировать ]- Колоректальный рак
- Полип (лекарство)
- Аденома
- Аденоматозные полипы
- Колоректальный полип
- Генетическое тестирование
Ссылки
[ редактировать ]- ^ Jump up to: а б с д и ж г Соравия, К.; Берк, Т.; Мадленский, Л.; Митри, А.; Ченг, Х.; Галлинджер, С.; Коэн, З.; Бапат, Б. (июнь 1998 г.). «Корреляции генотипа-фенотипа при ослабленном аденоматозном полипозе кишечной палочки» . Являюсь. Дж. Хум. Жене . 62 (6): 1290–1301. дои : 10.1086/301883 . ПМЦ 1377162 . ПМИД 9585611 .
- ^ Jump up to: а б с д и ж г GeneReviews NBK1345
- ^ Jump up to: а б «Семейный аденоматозный полипоз: MedlinePlus Genetics» . МедлайнПлюс . Проверено 9 июня 2023 г.
- ^ Баранов Е., Хорник Дж.Л. (март 2020 г.). «Специальный выпуск мягких тканей: фибробластические и миофибробластические новообразования головы и шеи» . Патология головы и шеи . 14 (1): 43–58. дои : 10.1007/s12105-019-01104-3 . ПМК 7021862 . ПМИД 31950474 .
- ^ Гарднер Э.Дж. (июнь 1951 г.). «Генетическое и клиническое исследование полипоза кишечника, предрасполагающего фактора рака толстой и прямой кишки» . Ам Джей Хум Жене . 3 (2): 167–76. ПМЦ 1716321 . ПМИД 14902760 .
- ^ Амос-Ландграф Дж., Квонг Л.Н., Дав В.Ф. и др. (2007). «Выбранная целевая группа крыс с мутацией Apc улучшает моделирование семейного рака толстой кишки человека» . Учеб. Натл. акад. наук. США . 104 (10): 4036–41. Бибкод : 2007PNAS..104.4036A . дои : 10.1073/pnas.0611690104 . ПМК 1805486 . ПМИД 17360473 .
- ^ Джардиелло Ф.М., Круш А.Дж., Петерсен ГМ, Букер С.В., Керр М., Тонг Л.Л., Гамильтон С.Р. (июнь 1994 г.). «Фенотипическая изменчивость семейного аденоматозного полипоза в 11 неродственных семьях с идентичной мутацией гена APC» . Гастроэнтерология . 106 (6): 1542–7. дои : 10.1016/0016-5085(94)90408-1 . ПМИД 8194700 .
- ^ Хес Ф.Дж., Нильсен М., Бик Э.К., Конвалинка Д., Вийнен Дж.Т., Баккер Э., Васен Х.Ф., Бройнинг М.Х., Топс СМ (январь 2008 г.). «Соматический мозаицизм APC: недооцененная причина полипоза кишечной палочки» . Гут . 57 (1): 71–6. дои : 10.1136/gut.2006.117796 . PMID 17604324 . S2CID 25082664 .
- ^ Линч, Патрик М; Берк, Кэрол А; Филлипс, Робин; Моррис, Джеффри С; Слэк, Ребекка; Ван, Сюэмэй; Лю, Цзюнь; Паттерсон, Шерри; Синикроп, Фрэнк А; Родригес-Бигас, Мигель А; Халф, Элизабет (февраль 2016 г.). «Международное рандомизированное исследование целекоксиба по сравнению с целекоксибом и дифторметилорнитином у пациентов с семейным аденоматозным полипозом» . Гут . 65 (2): 286–295. дои : 10.1136/gutjnl-2014-307235 . hdl : 10044/1/49491 . ISSN 0017-5749 . ПМИД 25792707 . S2CID 8059715 .
- ^ Берк, Кэрол А.; Деккер, Эвелен; Линч, Патрик; Самаддер, Н. Джуэл; Балагер, Франческ; Хюнебург, Роберт; Берн, Джон; Кастельс, Антони; Галлинджер, Стивен; Лим, Рамона; Стоффель, Елена М. (10 сентября 2020 г.). «Эфлорнитин плюс сулиндак для предотвращения прогрессирования семейного аденоматозного полипоза» . Медицинский журнал Новой Англии . 383 (11): 1028–1039. дои : 10.1056/NEJMoa1916063 . ISSN 0028-4793 . ПМИД 32905675 . S2CID 221620374 .
- ^ Берк, Кэрол А.; Деккер, Эвелен; Самаддер, Н. Джуэл; Стоффель, Елена; Коэн, Альфред (декабрь 2016 г.). «Эффективность и безопасность комбинированной терапии эфлорнитин (CPP-1X)/сулиндак по сравнению с каждым из них в качестве монотерапии у пациентов с семейным аденоматозным полипозом (САП): дизайн и обоснование рандомизированного двойного слепого исследования фазы III» . БМК Гастроэнтерология . 16 (1): 87. дои : 10.1186/s12876-016-0494-4 . ISSN 1471-230X . ПМЦ 4969736 . ПМИД 27480131 .
- ^ «Десмоидная опухоль – факторы риска» . Рак.Нет . 2020-09-02 . Проверено 19 августа 2023 г.
- ^ «Семейный аденоматозный полипоз» . Библиотека медицинских концепций Lecturio . Проверено 22 июля 2021 г.
- ^ Некласон, Дебора В.; Стивенс, Джеффри; Баучер, Кеннет М.; Кербер, Ричард А.; Мацунами, Нори; Барлоу, Ян; Мино, Джеральдин; Лепперт, Марк Ф.; Берт, Рэндалл В. (январь 2008 г.). «Американская мутация-основатель ослабленного семейного аденоматозного полипоза» . Клиническая гастроэнтерология и гепатология . 6 (1): 46–52. дои : 10.1016/j.cgh.2007.09.017 . ПМК 2245898 . ПМИД 18063416 .
- ^ Рейес Морено Дж., Джинард Висенс Д., Ванрел М. и др. (2007). «[Влияние реестра на выживаемость семейного аденоматозного полипоза.]». Клиническая медицина (на испанском языке). 129 (2): 51–2. дои : 10.1157/13106937 . ПМИД 17588361 .
Дальнейшее чтение
[ редактировать ]- Джасперсон, Кори В.; Патель, Свати Г.; Анен, Деннис Дж. (2 февраля 2017 г.). «Состояния полипоза, ассоциированного с APC» . В Адаме МП; Ардингер Х.Х.; Пагон РА; Уоллес С.Е. (ред.). Джин Обзоры . Сиэтл, Вашингтон: Вашингтонский университет. ПМИД 20301519 . НБК1345. - полное клиническое описание САП и ослабленного САП, включая пожизненные риски, эпидемиологию и т. д.
- Семейный аденоматозный полипоз в NLM Genetics домашнем справочнике
- Семейный аденоматозный полипоз — Электронная медицина Гастроэнтерология
- Синдромы толстой кишки, полипоза
- Национальный институт рака: сводная информация о генетике колоректального рака