Цинкорганическая химия
Цинкорганическая химия — это изучение физических свойств, синтеза и реакций цинкорганических соединений , которые представляют собой металлоорганические соединения , содержащие между углеродом (C) и цинком (Zn) химические связи . [1] [2] [3] [4]
Цинкорганические соединения были одними из первых металлоорганических соединений. Они менее реакционноспособны, чем многие другие аналогичные металлоорганические реагенты, такие как реактивы Гриньяра и литийорганические реактивы . В 1848 году Эдвард Франкленд получил первое цинкорганическое соединение диэтилцинк путем нагревания иодистого этила в присутствии металлического цинка. [5] В результате этой реакции образовалась летучая бесцветная жидкость, которая самопроизвольно воспламенялась при контакте с воздухом. Из-за своей пирофорной природы цинкорганические соединения обычно получают безвоздушным методом . Они неустойчивы по отношению к протонным растворителям . Для многих целей их готовят на месте , а не выделяют, но многие из них были выделены как чистые вещества и тщательно охарактеризованы. [6]
Цинкорганические соединения можно разделить на категории по количеству углеродных заместителей, связанных с металлом. [2] [3]
- Диорганоцинк ( R 2 Zn ): класс цинкорганических соединений, в которых имеются два алкильных лиганда. Их можно разделить на подклассы в зависимости от других лигандов . присоединенных
- Гетеролептические (RZnX): соединения, в которых электроотрицательный или моноанионный лиганд (X), например галогенид , присоединен к цинковому центру с помощью другого алкильного или арильного заместителя (R).
- Ионные цинкорганические соединения: Этот класс делится на цинкорганические соединения ( Р н Зн − ) и цинкорганические катионы ( РЗнЛ +
н ).
Склеивание
[ редактировать ]В своих координационных комплексах цинк (II) принимает несколько координационных геометрий, обычно октаэдрическую , тетраэдрическую и различные пентакоординатные геометрии. Эту структурную гибкость можно объяснить электронной конфигурацией цинка [Ar]3d. 10 4 с 2 . 3d-орбиталь заполнена, поэтому эффекты поля лигандов отсутствуют. Таким образом, координационная геометрия во многом определяется электростатическими и стерическими взаимодействиями. [2] Цинкорганические соединения обычно являются двух- или трехкоординатными, что отражает сильное донорное свойство карбанионных лигандов.
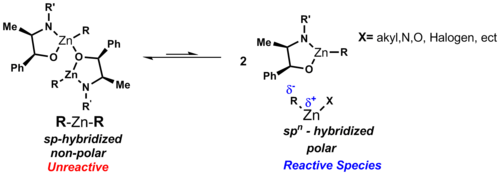
Типичные диорганоцинковые комплексы имеют формулу R 2 Zn. Диалкилцинковые соединения являются мономерами с линейной координацией у атома цинка. [7] Между углеродом и цинком существует полярная ковалентная связь , поляризованная в сторону углерода из-за различий в значениях электроотрицательности (углерод: 2,5 и цинк: 1,65). Дипольный момент симметричных диорганоцинковых реагентов в этих линейных комплексах можно рассматривать как нулевой, что объясняет их растворимость в неполярных растворителях, таких как циклогексан . В отличие от других бинарных алкилов металлов, диорганоцинковые соединения обладают низким сродством к комплексообразованию с эфирным растворителем. Связь в R 2 Zn описывается как использование sp- гибридных орбиталей на Zn. [2]
Эти структуры приводят к тому, что цинк имеет две связывающие d-орбитали и три низколежащие несвязывающие d-орбитали (см. несвязывающую орбиталь ), которые доступны для связывания. Когда у цинка отсутствуют электронодонорные лиганды, он не может получить координационное насыщение, что является следствием большого атомного радиуса и низкого дефицита электронов цинка. Следовательно, мостиковые алкильные или арильные группы возникают редко из-за слабого дефицита электронов атома цинка. Хотя в некоторых случаях это действительно происходит, например, когда Ph 2 Zn (показано ниже) и какие галогены являются цинкорганическими, они могут образовывать металлические кластеры (см. Химию кластеров ). Когда к атому цинка добавляется галогеновый лиганд, как акцепторный, так и донорный характер цинка усиливается, что позволяет осуществлять агрегацию. [2]

Синтез
[ редактировать ]Существует несколько методов получения цинкорганических соединений. Коммерчески доступными диорганоцинковыми соединениями являются диметилцинк , диэтилцинк и дифенилцинк. Эти реагенты дороги и сложны в обращении. В одном исследовании [8] [9] активное цинкорганическое соединение получают из гораздо более дешевых броморганических предшественников:
 | ( 2.1 ) |
Из металлического цинка
[ редактировать ]Франкландом Первоначальный синтез диэтилцинка включал реакцию этилиодида с металлическим цинком. Цинк необходимо активировать, чтобы облегчить эту окислительно-восстановительную реакцию. Одной из таких активированных форм цинка, используемых Франкландом, является пара цинк-медь . [5]
| 2 EtI + 2 Zn 0 → И 2 Zn + ZnI 2 | ( 2.2 ) |
Цинк Рике , получаемый восстановлением ZnCl 2 калием in situ, является еще одной активированной формой цинка. Эта форма оказалась полезной для таких реакций, как сочетание Негиши и сочетание Фукуямы . Образованию цинкорганических реагентов способствуют алкил- или арилгалогениды, содержащие электроноакцепторные заместители, например нитрилы и сложные эфиры. [10] [11]
| ( 2.3 ) |
![{\displaystyle {\ce {{ZnCl2}+2K->[{\ce {THF}}][{\ce {-2KCl}}]}}\overbrace {\ce {Zn^{0}}} ^{ \ce {Рикке\ цинк}}+{\ce {RX->[{\ce {THF}}][20-60^{\circ }{\ce {C}}]R-Zn-I}}\ qquad {\begin{cases}\mathbf {R} :& {\text{Аллил, арил, алкил, бензил}}\\\mathbf {X} :& {\text{Бромид, йодид}}\end{cases} }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/18c7cba23f482f24117a7664d614603a1271e9f2 "Цинк Reicke допускает использование активированных форм цинка для окислительного присоединения.")
 | ( 2.4 ) |
Обмен функциональными группами
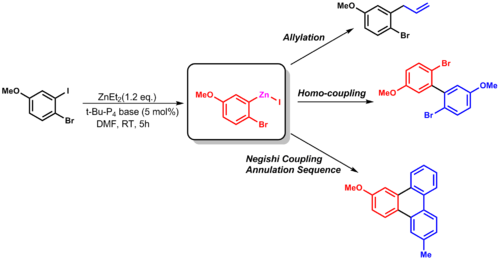
[ редактировать ]Двумя наиболее распространенными реакциями взаимного превращения функциональных групп цинка являются галогениды и бор, которые катализируются йодидом меди (CuI) или основанием. Промежуточное соединение бора синтезируют путем первоначальной реакции гидроборирования с последующей обработкой диэтилцинком . Этот синтез показывает полезность цинкорганических реагентов, демонстрируя высокую селективность в отношении наиболее реакционноспособного участка молекулы, а также создавая полезных партнеров для связывания. [12]
 | ( 2.5 ) |
Эту реакцию переноса группы можно использовать при аллилировании или других реакциях сочетания (например, связывании Негиши). [13]
 | ( 2.6 ) |
β-силилдиорганоцинковые соединения
[ редактировать ]Одним из основных недостатков диорганоцинкового алкилирования является перенос только одного из двух алкильных заместителей. Эту проблему можно решить, используя Me 3 SiCH 2 - (TMSM), который является непереносимой группой. [14]
| ( 2.7 ) |
![{\displaystyle {\begin{array}{l}{}\\{\ce {{R2Zn}+(TMSM)2Zn}} \ {\overset {\ce {THF}}{\ce {<=>>} }}\ {\ce {2R(TMSM)Zn}}\\{}\\{\ce {{RZnI}+(TMSM)Li->[{\ce {THF}}][-80^{\circ }\!{\ce {C}}]{R(TMSM)Zn}+LiI}}\\{}\end{array}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0fdd1941ffd29949716def994df7c380dd816d96 "Кнохель и др. Добавление переноса бета-силильной группы")
Трансметаллизация
[ редактировать ]Трансметаллирование аналогично взаимопревращениям, показанным выше, цинк может обмениваться с другими металлами, такими как ртуть , литий , медь и т. д. Одним из примеров этой реакции является реакция дифенилртути с металлическим цинком с образованием дифенилцинка и металлической ртути :
| HgPh 2 + Zn → ZnPh 2 + Hg | ( 2.8 ) |
Преимущество трансметаллирования в цинк часто заключается в большей толерантности к другим функциональным группам в молекуле из-за низкой реакционной способности, которая увеличивает селективность. [15]
- При синтезе Маокристалла V направленное орто-металлирование дает исходную разновидность ариллития, которая трансметаллируется до желаемого соединения арилцинка. Соединение арилцинка значительно менее реакционноспособно, чем соединение ариллития, и поэтому лучше переносит функциональность при последующем сочетании с метилхлороксалоацетатом. Эфиры значительно устойчивы к цинкорганическим реагентам. [16]
 | ( 2.9 ) |
Органоцинк можно получить непосредственно из металлического цинка: [17] [18]
 | ( 2.10 ) |
- В этом методе цинк активируется 1,2-дибромэтаном и триметилсилилхлоридом . Ключевым ингредиентом является хлорид лития , который быстро образует растворимый аддукт с цинкорганическим соединением, удаляя его с поверхности металла.
Реакции
[ редактировать ]Во многих их реакциях цинкорганические соединения выступают в качестве промежуточных продуктов.
- В реакции Франкланда-Дуппы (1863 г.) эфир оксалата (ROCOCOOR) реагирует с алкилгалогенидом R'X, цинком и соляной кислотой с образованием сложных эфиров α-гидроксикарбоновых кислот RR'COHCOOR. [19]
Реформатская реакция
[ редактировать ]Эту органическую реакцию можно использовать для превращения α-галогенэфира и кетона или альдегида в β-гидроксиэфир. Кислота необходима для протонирования образующегося алкоксида во время обработки. Начальным этапом является окислительное присоединение металлического цинка к связи углерод-галоген с образованием енолята углерод-цинк. C-Zn Этот енолят может затем посредством координации перегруппироваться в енолят кислорода-цинка. Как только он образуется, другой исходный материал, содержащий карбонил, будет координироваться, как показано ниже, и давать продукт после протонирования. [20] Преимущества реакции Реформатского по сравнению с традиционными протоколами альдольных реакций заключаются в следующем:
- Позволяет использовать чрезвычайно дериватизированные кетоновые субстраты.
- сложного эфира Промежуточный енолят может быть образован в присутствии енолизируемых фрагментов.
- Хорошо подходит для внутримолекулярных реакций.
Ниже показано шестичленное переходное состояние модели Циммермана-Тракслера (Контроль хелатирования, см. Реакцию Альдола ), в котором R 3 меньше, чем R 4 . [21]
 | ( 3.1 ) |
Реакция Реформатского использовалась во многих полных синтезах, таких как синтез C(16),C(18)-бис-эпицитохалазина D: [22]
 | ( 3.2 ) |
Реакция Реформатского допускает даже использование гомоенолятов цинка. [23] Модификацией реакции Реформатского является реакция Блеза . [21]
 | ( 3.3 ) |
Реакция Симмонса-Смита
[ редактировать ]Реагент Симмонса -Смита используется для получения циклопропанов из олефинов с использованием иодистого метилена в качестве источника метилена. Реакцию осуществляют с цинком. Ключевым образующимся промежуточным соединением цинка является карбеноид (иодметил) йодид цинка, который реагирует с алкенами с образованием циклопропанированного продукта. Скорость образования активных форм цинка увеличивается за счет обработки ультразвуком, поскольку первоначальная реакция происходит на поверхности металла.
| ( 3.4 ) |

 | ( 3.5 ) |
Хотя механизм до конца не изучен, предполагается, что цинкорганическое промежуточное соединение представляет собой металл- карбеноид . Считается, что промежуточное соединение представляет собой трехцентровую структуру типа «бабочка». Это промежуточное соединение может быть направлено заместителями, такими как спирты, для доставки циклопропана на одну и ту же сторону молекулы. Пара цинк-медь обычно используется для активации цинка. [21]
 | ( 3.6 ) |
Метилиденирование титана-цинка
[ редактировать ]Цинкорганические соединения, производные бромистого или иодида метилена, могут электрофильно присоединяться к карбонильным группам с образованием концевых алкенов . [24] Реакция механически связана с реакцией Теббе и может катализироваться различными кислотами Льюиса (например, TiCl 4 или Al 2 Me 6 ). [25] Реакция используется для введения дейтерия в молекулы для изотопной маркировки или в качестве альтернативы реакции Виттига .
Муфта Негиши
[ редактировать ]Эта мощная углерод-углеродная связь, образующая реакции кросс-сочетания, объединяет органический галогенид и цинкорганический галогенид в присутствии никелевого или палладиевого катализатора . Органический галоидный реагент может представлять собой алкенил , арил , аллил или пропаргил . Сообщалось также о сочетании алкилцинка с алкилгалогенидами, такими как бромиды и хлориды, с использованием активных катализаторов, таких как предкатализаторы Pd-PEPPSI, которые сильно сопротивляются удалению бета-гидрида (обычное явление с алкильными заместителями). [26] Либо диорганический [ проверьте орфографию ] виды или цинкорганические галогениды могут использоваться в качестве партнеров сочетания на стадии трансметаллирования в этой реакции. Несмотря на низкую реакционную способность цинкорганических реагентов по отношению к органическим электрофилам, эти реагенты относятся к числу наиболее сильных металлонуклеофилов по отношению к палладию. [27]
Разновидности алкилцинка требуют присутствия по крайней мере стехиометрического количества галогенид-ионов в растворе для образования «цинкатных» разновидностей формы RZnX 3. 2− , прежде чем он сможет подвергнуться трансметаллизации в палладиевый центр. [28] Такое поведение сильно контрастирует со случаем арилцинковых форм. Ключевым этапом каталитического цикла является трансметаллирование , при котором галогенид цинка обменивает свой органический заместитель на другой галоген с металлическим центром.
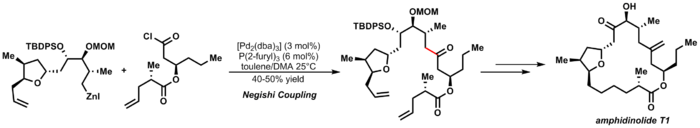
Элегантным примером сочетания Негиши является синтез Фюрстнером амфидинолида Т1: [29]
 | ( 3.7 ) |
Муфта Фукуяма
[ редактировать ]Сочетание Фукуямы представляет собой реакцию, катализируемую палладием, включающую сочетание арильного, алкильного, аллильного или α,β-ненасыщенного тиоэфирного соединения. Это тиоэфирное соединение можно сочетать с широким спектром цинкорганических реагентов, чтобы выявить соответствующий кетоновый продукт. Этот протокол полезен из-за его чувствительности к функциональным группам, таким как кетон , ацетат , ароматические галогениды и даже альдегиды. Наблюдаемая хемоселективность указывает на то, что образование кетонов происходит более легко, чем окислительное присоединение палладия к этим другим фрагментам. [30]
 | ( 3.8 ) |
Еще одним примером этого метода сочетания является синтез (+)- биотина . В этом случае происходит реакция Фукуямы с тиолактоном: [31]
 | ( 3.9 ) |
Реакция Барбье
[ редактировать ]Реакция Барбье включает нуклеофильное присоединение карбаниона, эквивалентного карбонилу. Превращение аналогично реакции Гриньяра. Цинкорганический реагент получают путем окислительного присоединения к алкилгалогениду. В результате реакции образуется первичный, вторичный или третичный спирт посредством 1,2-присоединения . Реакция Барбье выгодна тем, что это однореакторный процесс: цинкорганический реагент образуется в присутствии карбонильного субстрата. Цинкорганические реагенты также менее чувствительны к воде, поэтому эту реакцию можно проводить в воде. Подобно реакции Гриньяра, применяется равновесие Шленка , при котором может образовываться более реакционноспособный диалкилцинк. [21]
 | ( 3.10 ) |
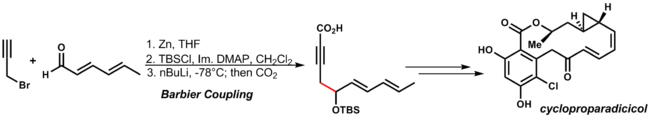
Механизм напоминает реакцию Гриньяра , в которой алкоксид металла может генерироваться радикальным ступенчатым путем, посредством переноса одного электрона , или согласованным путем реакции через циклическое переходное состояние. Примером этой реакции является . синтез циклопропарадицикола Данишевским При использовании условий реакции цинкорганического присоединения допускаются другие функциональные группы диенона и алкина: [32]
 | ( 3.11 ) |
Ацетилиды цинка
[ редактировать ]Образование ацетилида цинка происходит через посредство диалкинила цинка (обмен функциональных групп). Были разработаны каталитические процессы, такие как эфедриновый процесс компании Merck. [33] Пропаргиловые спирты можно синтезировать из ацетилидов цинка. Эти универсальные промежуточные соединения затем можно использовать для широкого спектра химических превращений, таких как реакции кросс-сочетания , гидрирования и перициклические реакции . [34]
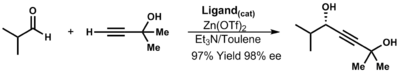 | ( 3.12 ) |
В отсутствие лигандов реакция протекает медленно и неэффективно. В присутствии хиральных лигандов реакция протекает быстро и дает высокую конверсию. Рёдзи Нойори определил, что комплекс моноцинк-лиганд является активной разновидностью. [35]
 | ( 3.13 ) |
Диастереоселективность присоединения цинкорганических реагентов к альдегидам можно предсказать с помощью следующей модели Нойори и Дэвида А. Эванса : [36]
 | ( 3.14 ) |
- α- стереоцентр лиганда определяет наблюдаемую стереохимию пропаргиловых спиртов.
- Стерические эффекты между альдегидным заместителем и лигандом менее важны, но все же определяют предпочтительную конформацию.
Ацетилиды цинка используются в ВИЧ-1 обратной транскриптазы ингибиторе эфавиренц, а также в производных эфедрина компании Merck . [37]
 | ( 3.15 ) |
Органоцинкаты
[ редактировать ]О первом цинкорганическом комплексе ( цинкорганическом комплексе ) сообщил в 1858 году Джеймс Альфред Ванклин . [38] помощник Франкланда и касался реакции элементарного натрия с диэтилцинком :
| 2 Na + 3 ZnEt 2 → 2 NaZnEt 3 + Zn | ( 4.1 ) |
Цинкорганические соединения, которые являются сильными кислотами Льюиса , уязвимы для нуклеофильной атаки щелочных металлов , таких как натрий , и, таким образом, образуют эти «атомные соединения». Выделяют два типа цинкорганических соединений: тетраорганоцинкаты ([R 4 Zn]M 2 ), которые являются дианионными, и триорганоцинкаты ([R 3 Zn]M), которые являются моноанионными. Их структуры, определяемые лигандами, тщательно охарактеризованы. [3]
Синтез
[ редактировать ]Тетраорганоцинкаты, такие как [Me 4 Zn]Li 2, можно получить путем смешивания Me 2 Zn и MeLi в молярном соотношении ректантов 1:2. Другой пример синтетического пути образования спиоциклических органоцинкатов показан ниже: [3]
 | ( 4.2 ) |
Триорганоцинкатные соединения образуются путем обработки диорганоцинка, такого как (Me 3 SiCH 2 ) 2 Zn, щелочным металлом (K) или щелочноземельным металлом (Ba, Sr или Ca). Одним из примеров является [(Me 3 SiCH 2 ) 3 Zn]K.Триэтилцинкат разлагается до гидридоэтилцинката(II) натрия в результате отщепления бета-гидрида : [39]
| 2 NaZnEt 3 → Na 2 Zn 2 H 2 Et 4 + 2 C 2 H 4 | ( 4.3 ) |
Продукт представляет собой битетраэдрическую структуру с общими краями и мостиковыми гидридными лигандами .
Реакции
[ редактировать ]Хотя цинкорганические соединения изучаются реже, они часто обладают повышенной реакционной способностью и селективностью по сравнению с нейтральными диорганоцинковыми соединениями. Они были полезны в стереоселективном алкилировании кетонов и родственных карбонилов, реакциях раскрытия кольца. Арилтриметилцинкаты участвуют в реакциях образования CC, опосредованных ванадием. [3]
 | ( 4.4 ) |
Цинкорганические(I) соединения
[ редактировать ]Известны также низковалентные цинкорганические соединения, имеющие связь Zn–Zn. О первом таком соединении, декаметилдизинкоцене , сообщалось в 2004 году. [40]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Кнохель, Пол; Милло, Николя; Родригес, Ален Л.; Такер, Чарльз Э. (2004). Органические реакции . дои : 10.1002/0471264180.или058.02 . ISBN 0471264180 .
- ^ Jump up to: а б с д и Химия цинкорганических соединений (Серия Патаи: Химия функциональных групп ), (ред. З. Раппопорт и И. Марек), John Wiley & Sons: Чичестер, Великобритания, 2006 г. , ISBN 0-470-09337-4 .
- ^ Jump up to: а б с д и Цинкорганические реагенты – практический подход (под ред. П. Кнохеля и П. Джонса), Oxford Medical Publications, Оксфорд, 1999 г. , ISBN 0-19-850121-8 .
- ^ Синтетические методы металлоорганической и неорганической химии, том 5, Медь, серебро, золото, цинк, кадмий и ртуть , WA Herrmann Ed., ISBN 3-13-103061-5
- ^ Jump up to: а б Э. Франкланд, Либигс Энн. хим., 1849, 71, 171.
- ^ Эльшенбройх, К. «Металлоорганические соединения» (2006) Wiley-VCH: Weinheim. ISBN 978-3-527-29390-2
- ^ Джон Бакса; Феликс Ханке; Сара Хиндли; Раджеш Одедра; Джордж Р. Дарлинг; Энтони С. Джонс; Александр Штайнер (2011). «Твердотельные структуры диметилцинка и диэтилцинка» . Angewandte Chemie, международное издание . 50 (49): 11685–11687. дои : 10.1002/anie.201105099 . ПМЦ 3326375 . ПМИД 21919175 .
- ^ Ким, Юнг Гон; Уолш, Патрик Дж. (2006). «От арилбромидов к энантиообогащенным бензиловым спиртам в одной колбе: каталитическое асимметричное арилирование альдегидов» . Angewandte Chemie, международное издание . 45 (25): 4175–4178. дои : 10.1002/anie.200600741 . ПМИД 16721894 .
- ^ В этой реакции в одном котле бромбензол превращается в фениллитий путем реакции с 4 эквивалентами н -бутиллития , затем трансметаллирование с хлоридом цинка образует дифенилцинк, который продолжает реагировать в асимметричной реакции сначала с лигандом MIB , а затем с 2-нафтилальдегидом до алкоголь . В этой реакции образование дифенилцинка сопровождается образованием хлорида лития , который бесконтрольно катализирует реакцию без участия МИБ с рацемическим спиртом. Соль эффективно удаляется путем хелатирования тетраэтилендиамином энантиомерному (TEEDA), что приводит к избытку 92%.
- ^ Рике, Р.Д. (1989). «Получение металлоорганических соединений из высокореактивных металлических порошков». Наука . 246 (4935): 1260–1264. Бибкод : 1989Sci...246.1260R . дои : 10.1126/science.246.4935.1260 . ПМИД 17832221 . S2CID 92794 .
- ^ Негиси, Эй-Ичи (2002). «Генеалогия кросс-сочетания, катализируемого Pd». Журнал металлоорганической химии . 653 (1–2): 34–40. дои : 10.1016/S0022-328X(02)01273-1 .
- ^ Лангер, Фальк; Швинк, Лотар; Девасагаярадж, Арокиасами; Шаван, Пьер-Ив; Кнохель, Пол (1996). «Получение функционализированных диалкилцинков посредством бор-цинкового обмена. Реакционная способность и каталитическое асимметрическое присоединение к альдегидам». Журнал органической химии . 61 (23): 8229–8243. дои : 10.1021/jo961129n . ISSN 0022-3263 . PMID 11667810 .
- ^ Naka,H; et al.New J. Chem., 2010, 34, 1700–1706
- ^ Knochel,P.; et al. Angel. Chem. Int. Ed. Engl. 1997, volume 36, 1496-1498
- ^ Маркис, П; Шат, Геррит; Аккерман, Отто С.; Бикельхаупт, Ф.; Спек, Энтони Л. (1992). «Комплексообразование дифенилцинка с простыми эфирами. Кристаллические структуры комплексов Ph 2 Zn·глим и Ph2Zn·диглим». Дж. Органомет. хим. 430 : 1–13. дои : 10.1016/0022-328X(92)80090-K .
- ^ Лу, Пин; Гу, Чжэньхуа; Закарян, Армен (2013). «Полный синтез маокристалла V: ранняя стадия функционализации C – H и сборка лактона посредством радикальной циклизации» . Журнал Американского химического общества . 135 (39): 14552–5. дои : 10.1021/ja408231t . ПМК 4118676 . ПМИД 24047444 .
- ^ Красовский, Аркадий; Малахов Владимир; Гаврюшин Андрей; Кнохель, Пол (2006). «Эффективный синтез функционализированных цинкорганических соединений путем прямого введения цинка в органические йодиды и бромиды». Angewandte Chemie, международное издание . 45 (36): 6040–6044. дои : 10.1002/anie.200601450 . ПМИД 16900548 .
- ^ В этом примере арилцинк иодид продолжает реагировать с аллилбромидом в результате нуклеофильного замещения.
- ^ Индекс Мерк . Merck & Co. 2001. ISBN. 9780911910131 . Реакция Франкланда-Дуппы.
- ^ Фюрстнер, Алоис (1989). «Последние достижения в реакции Реформатского». Синтез . 1989 (8): 571–590. дои : 10.1055/s-1989-27326 . S2CID 94339252 . )
- ^ Jump up to: а б с д Курти, Л.; Чако, Б. Стратегическое применение названных реакций в органическом синтезе ; Эльзевир: Берлингтон, 2005.
- ^ Ведейс, Э.; Дункан, С.М. (2000). «Синтез C (16), C (18)-бис-эпицитохалазина D посредством циклизации Реформатского». Журнал органической химии . 65 (19): 6073–81. дои : 10.1021/jo000533q . ПМИД 10987942 .
- ^ Погребение, И.; и др. Дж. Ам. хим. 1987, 109, 8056
- ^ Такай, Кадзухико; Хотта, Юджи; Осима, Коитиро; Нодзаки, Хитоси (1980). «Реакция типа Виттига диметаллированных карбодианионов, полученная восстановлением цинка гем-полигалогенных соединений в присутствии кислот Льюиса» . Бюллетень Химического общества Японии . 53 (6): 1698–1702. дои : 10.1246/bcsj.53.1698 .
- ^ Трост, Барри; Флеминг, Ян; Шрайбер, Стюарт (1991). «Превращение карбонильной группы в негидроксильные группы». Комплексный органический синтез, том 1: Дополнения к CX π-связям, часть 1 (1-е изд.). Нью-Йорк: Пергамон Пресс. стр. 749–751. дои : 10.1016/B978-0-08-052349-1.00020-2 . ISBN 9780080405926 .
- ^ S. Sase, M. Jaric, A. Metzger, V. Malakhov, P. Knochel, J. Org. Chem., 2008, 73, 7380-7382
- ^ Николау, КЦ; Балджер, Пол Г.; Сарла, Дэвид (2005). «Реакции кросс-сочетания, катализируемые палладием, в полном синтезе». Angewandte Chemie, международное издание . 44 (29): 4442–4489. дои : 10.1002/anie.200500368 . ПМИД 15991198 . )
- ^ Макканн, ЖК; Хантер, Х.Н.; Сайберн, JAC; Орган, MG (2012). «Цинкаты высшего порядка как трансметаллаторы в перекрестном сочетании алкил-алкил Негиши». Энджью. хим. Межд. Эд . 51 (28): 7024–7027. дои : 10.1002/anie.201203547 . ПМИД 22685029 .
- ^ Аисса, Кристоф; Ривейрос, Рикардо; Раго, Жак; Фюрстнер, Алоис (2003). «Полный синтез амфидинолидов Т1, Т3, Т4 и Т5». Журнал Американского химического общества . 125 (50): 15512–20. дои : 10.1021/ja038216z . ПМИД 14664598 .
- ^ Токуяма, Хидэтоси; Йокосима, Сатоши; Ямасита, Тору; Фукуяма, Тору (1998). «Новый синтез кетонов катализируемой палладием реакцией тиоловых эфиров и цинкорганических реагентов». Буквы тетраэдра . 39 (20): 3189–3192. дои : 10.1016/S0040-4039(98)00456-0 .
- ^ Симидзу, Тошиаки; Секи, Масахико (2000). «Легкий синтез (+)-биотина посредством реакции сочетания Фукуямы». Буквы тетраэдра . 41 (26): 5099–5101. дои : 10.1016/S0040-4039(00)00781-4 .
- ^ Ян, Чжи-Цян; Гэн, Сюйдун; Солит, Дэвид; Пратилас, Кристина А.; Розен, Нил; Данишефски, Сэмюэл Дж. (2004). «Новый эффективный синтез резорциниловых макролидов с помощью инолидов: создание циклопропарадицикола в качестве синтетически возможного доклинического противоракового агента на основе Hsp90 в качестве мишени». Журнал Американского химического общества . 126 (25): 7881–9. дои : 10.1021/ja0484348 . ПМИД 15212536 .
- ^ Ли, З.; Упадхьяй, В.; ДеКамп, А.Е.; ДиМишель, Л.; Рейдер, П.Дж. Синтез 1999, 1453–1458.
- ^ Соаи, Кенсо; Нива, Сейджи (1992). «Энантиоселективное присоединение цинкорганических реагентов к альдегидам». Химические обзоры . 92 (5): 833–856. дои : 10.1021/cr00013a004 .
- ^ Ноёри, Рёдзи; Китамура, Масато (1991). «Энантиоселективное добавление металлоорганических реагентов к карбонильным соединениям: перенос хиральности, умножение и амплификация». Angewandte Chemie International Edition на английском языке . 30 : 49–69. дои : 10.1002/anie.199100491 .
- ^ Эванс, Д. (1988). «Стереоселективные органические реакции: катализаторы процессов присоединения карбонила». Наука . 240 (4851): 420–6. Бибкод : 1988Sci...240..420E . дои : 10.1126/science.3358127 . ПМИД 3358127 .
- ^ Томпсон, AS; Корли, Е.Г.; Хантингтон, МФ; Грабовски, EJJ Tetrahedron Lett. 1995, 36, 8937-8940
- ^ Дж. А. Ванклин (1858). «О некоторых новых этиловых соединениях, содержащих щелочные металлы» . Анналы Либиха . 108 (67): 67–79. дои : 10.1002/jlac.18581080116 .
- ^ Леннартсон, Андерс; Хоканссон, Микаэль; Ягнер, Сьюзен (2007). «Простой синтез четко определенных гидридоалкилцинкатов натрия (II)». Angewandte Chemie, международное издание . 46 (35): 6678–6680. дои : 10.1002/anie.200701477 . ПМИД 17665387 .
- ^ Шульц, Стефан (2010). «Низковалентные металлоорганические соединения: синтез, реакционная способность и потенциальные применения» (PDF) . Химия: Европейский журнал . 16 (22): 6416–28. дои : 10.1002/chem.201000580 . ПМИД 20486240 .
Внешние ссылки
[ редактировать ]Соединения углерода с другими элементами таблицы Менделеева | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Легенда |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||