Эпигенетика нейродегенеративных заболеваний
Эта статья нуждается в более надежных медицинских ссылках для проверки или слишком сильно полагается на первоисточники . ( май 2015 г. ) |  |
Нейродегенеративные заболевания представляют собой гетерогенную группу сложных заболеваний, связанных с дегенерацией нейронов периферической системы или центральной нервной . Их основные причины чрезвычайно разнообразны и осложняются различными генетическими факторами и/или факторами окружающей среды. Эти заболевания вызывают прогрессирующее ухудшение состояния нейрона, что приводит к снижению передачи сигнала , а в некоторых случаях даже к гибели нейронов. Заболевания периферической нервной системы можно дополнительно классифицировать по типу нервных клеток ( моторные , сенсорные или оба), пораженных заболеванием. Эффективному лечению этих заболеваний часто препятствует отсутствие понимания лежащей в их основе молекулярной и генетической патологии. Эпигенетическая терапия исследуется как метод коррекции уровня экспрессии неправильно регулируемых генов при нейродегенеративных заболеваниях.
Нейродегенеративные заболевания мотонейронов могут вызывать дегенерацию мотонейронов, участвующих в произвольном мышечном контроле, таком как сокращение и расслабление мышц. В этой статье будут рассмотрены эпигенетика и лечение бокового амиотрофического склероза (БАС) и спинальной мышечной атрофии (СМА). См. информационный бюллетень по двигательным нейронам. [1] для получения подробной информации о других заболеваниях двигательных нейронов. Нейродегенеративные заболевания центральной нервной системы могут поражать головной и спинной мозг . В этой статье будут рассмотрены эпигенетика и лечение болезни Альцгеймера (БА), болезни Хантингтона (БГ) и болезни Паркинсона (БП). Эти заболевания характеризуются хронической и прогрессирующей нейрональной дисфункцией, иногда приводящей к поведенческим отклонениям (как при БП) и, в конечном итоге, к гибели нейронов, приводящей к деменции .
Нейродегенеративные заболевания сенсорных нейронов могут вызывать дегенерацию сенсорных нейронов, участвующих в передаче сенсорной информации, например, в слухе и зрении . Основной группой заболеваний сенсорных нейронов являются наследственные сенсорные и вегетативные невропатии (HSAN), такие как HSAN I , HSAN II и Шарко-Мари-Тута (CMT2B). тип 2B [2] [3] Хотя некоторые заболевания сенсорных нейронов признаны нейродегенеративными, эпигенетические факторы в молекулярной патологии еще не выяснены.
Эпигенетика и эпигенетические препараты
[ редактировать ]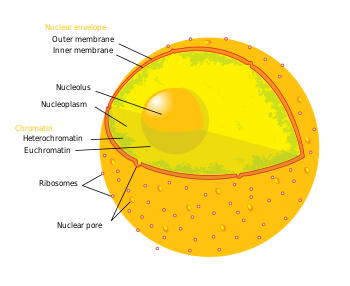
Термин «эпигенетика» относится к трем уровням регуляции генов: (1) метилирование ДНК , (2) модификации гистонов и (3) функция некодирующей РНК (нкРНК). Коротко говоря, опосредованный гистонами контроль транскрипции происходит путем обертывания ДНК вокруг ядра гистона . Эта структура ДНК-гистонов называется нуклеосомой ; чем прочнее ДНК связана с нуклеосомой и чем сильнее сжата между собой цепочка нуклеосом, тем сильнее репрессивный эффект на транскрипцию генов в последовательностях ДНК, расположенных рядом с гистонами или обернутых вокруг них, и наоборот (т.е. более слабое связывание ДНК и расслабленное уплотнение приводят к сравнительно дерепрессированному состоянию, что приводит к факультативному гетерохроматину или, еще более дерепрессированному, эухроматину ). В наиболее репрессивном состоянии, включающем в себя множество складок и другие каркасные белки, ДНК-гистоновые структуры образуют конститутивный гетерохроматин. Эта структура хроматина опосредована этими тремя уровнями регуляции генов. Наиболее важными эпигенетическими модификациями для лечения нейродегенеративных заболеваний являются метилирование ДНК и модификации белков-гистонов посредством метилирования или ацетилирования. [4] [5]
- У млекопитающих метилирование происходит на ДНК и белках-гистонах. Метилирование ДНК происходит на цитозине динуклеотидов CpG в геномной последовательности, а метилирование белка происходит на аминоконцах основных белков-гистонов – чаще всего на остатках лизина. [5] CpG относится к динуклеотиду, состоящему из цитозиндезоксинуклеотида, непосредственно примыкающего к гуаниндезоксинуклеотиду. Кластер CpG-динуклеотидов, сгруппированных вместе, называется CpG-островком , и у млекопитающих эти CpG-островки являются одним из основных классов генных промоторов, на которых или вокруг которых могут связываться транскрипционные факторы и может начаться транскрипция. Метилирование динуклеотидов и/или островков CpG в промоторах генов связано с репрессией транскрипции посредством вмешательства в связывание факторов транскрипции и рекрутирования репрессоров транскрипции с метилсвязывающими доменами. Метилирование внутригенных областей связано с усилением транскрипции. Группа ферментов, ответственных за добавление метильных групп к ДНК, называется ДНК-метилтрансферазами (ДНМТ). Ферменты, ответственные за удаление метильной группы, называются ДНК-деметилазами. Эффекты метилирования гистонов зависят от остатка (например, какая аминокислота на каком хвосте гистона метилируется), поэтому результирующая транскрипционная активность и регуляция хроматина может варьироваться. [5] Ферменты, ответственные за добавление метильных групп к гистонам, называются гистон-метилтрансферазами (HMT). Ферменты, ответственные за удаление метильных групп из гистона, — это деметилазы гистонов .
- Ацетилирование происходит по остаткам лизина, обнаруженным на амино-N-конце хвостов гистонов. Ацетилирование гистонов чаще всего связано с расслаблением хроматина, дерепрессией транскрипции и, следовательно, с активной транскрипцией генов. [5] Гистоновые ацетилтрансферазы (HAT) представляют собой ферменты, ответственные за добавление ацетильных групп, а гистондеацетилазы (HDAC) — ферменты, ответственные за удаление ацетильных групп. Следовательно, добавление или удаление ацетильной группы к гистону может изменить экспрессию близлежащих генов. Большинство исследуемых препаратов являются ингибиторами белков, удаляющих ацетил из гистонов, или гистондеацетилаз (HDAC).
- Вкратце, нкРНК участвуют в сигнальных каскадах с помощью эпигенетических маркирующих ферментов, таких как HMT, и/или с механизмом РНК-интерференции (RNAi). Часто эти сигнальные каскады приводят к эпигенетической репрессии (например, см. « Инактивация Х-хромосомы» ), хотя в некоторых случаях верно обратное. Например, экспрессия нкРНК BACE1-AS повышается у пациентов с болезнью Альцгеймера и приводит к повышению стабильности BACE1 – предшественника мРНК фермента, участвующего в болезни Альцгеймера. [6]
Эпигенетические препараты нацелены на белки, ответственные за модификации ДНК или гистонов. Современные эпигенетические препараты включают, помимо прочего: ингибиторы HDAC (HDACi), модуляторы HAT, ингибиторы ДНК-метилтрансферазы и ингибиторы деметилазы гистонов. [7] [8] Большинство эпигенетических препаратов, протестированных для применения против нейродегенеративных заболеваний, представляют собой ингибиторы HDAC; однако были также протестированы некоторые ингибиторы DNMT. Хотя большинство эпигенетических лекарственных препаратов проводилось на мышах, некоторые эксперименты проводились на человеческих клетках, а также в испытаниях лекарств на людях (см. таблицу ниже). Существуют неизбежные риски при использовании эпигенетических препаратов в качестве терапии нейродегенеративных расстройств, поскольку некоторые эпигенетические препараты (например, HDAC, такие как бутират натрия ) неспецифичны в своих мишенях, что оставляет возможность появления нецелевых эпигенетических меток, вызывающих нежелательные эпигенетические модификации.
| Функция | Классификация | Лекарство | ЕСЛИ | ОБЪЯВЛЕНИЕ | HD | ПД | СМА |
|---|---|---|---|---|---|---|---|
| Ингибитор метилирования ДНК | химический цитидина аналог | Азатиоприн | М (те) | М (те) | |||
| Ингибитор HDAC ( малая молекула ) | бензамид | М344 | МК 19 | ||||
| жирная кислота | Бутират натрия | М(у) 5, 6, 7 ; Н (или) | Д(г) 11 | М(у) 14 ; Р(у) 15 ; Д(у) 16, 18 ; Н (или) | МК 20 ; М(у) 21 ; Н (или) | ||
| Фенилбутират натрия | М(у) 1 ; Ч(у) 2 | М(у) 8 ; Н (или) | Ч (д) 12 | МК 20 ; Ч(в) 21, 22 | |||
| Вальпроевая кислота | М(у) 2 ; Х(ни) 3 | М(у) 9 ; Н (или) | Д(г) 11 | Р(у) 17 ; Н (или) | МЦ 23, 24 ; М(у) 25 ; Ч (в) 26, 27, 28, 29 | ||
| гидроксамовая кислота | Трихостатин А | М (у) 4 ; Н (или) | М (у) 10 ; Н (или) | МК 13 ; Д(г) 11 | М(у) 30, 31 ; Н (или) | ||
| Вориностат ( суберанилогидроксамовая кислота -SAHA) | М(у) 9 ; Н (или) | МК 13 ; Д(г) 11 | Д(г) 18 | МК 32, 33 ; М(у) 34 ; Н (или) |
- Заболевания: боковой амиотрофический склероз (БАС), болезнь Альцгеймера (БА), болезнь Хантингтона (БГ), спинальная мышечная атрофия (СМА), болезнь Паркинсона (БП).
- Тестировано на: мышах (М), только клетках мыши (MC), человеке (H), дрозофиле (D), крысе (R)
- Успешное лечение: да (да), да, но с побочными эффектами (да), пока нет (нет), варьируется (в), нет улучшения (нет)
- Ссылки: перечислены в столбцах (заболевания) и в порядке возрастания строк (препараты).
- ЕСЛИ : (1) [9] [10] (2) [11] (3) [12] (4) [13]
- А.Д .: (5) [14] (6) [15] (7) [16] (8) [15] (9) [17] (10) [18]
- HD : (11) [19] (12) [20] (13) [21]
- ПД : (14) [22] (15) [23] (16) [24] (17) [25] (18) [26]
- СМА : (19) [27] (20) [28] (21) [29] (22) [30] (23) [31] (24) [32] (25) [33] (26) [34] (27) [35] (28) [36] (29) [37] (30) [38] (31) [39] (32) [40] (33) [41] (34) [42]
Нейродегенеративные заболевания двигательных нейронов
[ редактировать ]Боковой амиотрофический склероз (АЛС)
[ редактировать ]Боковой амиотрофический склероз (БАС), также известный как болезнь Лу Герига, представляет собой заболевание двигательных нейронов, которое включает нейрогенерацию. Все скелетные мышцы в организме контролируются мотонейронами, которые передают сигналы от мозга к мышцам через нервно-мышечные соединения . Когда мотонейроны деградируют, мышцы перестают получать сигналы от мозга и начинают угасать. БАС характеризуется жесткостью мышц, мышечными подергиваниями и прогрессирующей мышечной слабостью из-за мышечного истощения. Части тела, пораженные ранними симптомами БАС, зависят от того, какие мотонейроны в организме повреждаются первыми, обычно это конечности. По мере прогрессирования заболевания большинство пациентов не могут ходить или пользоваться руками, и со временем у них возникают трудности с речью, глотанием и дыханием. У большинства пациентов когнитивные функции сохраняются, а сенсорные нейроны обычно не поражаются. Пациентам часто ставят диагноз после 40 лет, а среднее время выживания от начала заболевания до смерти составляет около 3–4 лет. На последних стадиях пациенты могут потерять произвольный контроль над глазными мышцами и часто умирают от дыхательная недостаточность или пневмония в результате дегенерации двигательных нейронов и мышц, необходимых для дыхания. В настоящее время не существует лекарства от БАС, есть только методы лечения, которые могут продлить жизнь.
Генетика и основные причины
[ редактировать ]На сегодняшний день в развитии БАС задействовано множество генов и белков. Одной из общих тем между многими из этих генов и вызывающими их мутациями является наличие белковых агрегатов в мотонейронах. [43] Другими общими молекулярными особенностями у пациентов с БАС являются измененный метаболизм РНК. [44] и общее гипоацетилирование гистонов. [45]
- СОД1
- Ген SOD1 на 21 хромосоме , который кодирует белок супероксиддисмутазы, связан с 2% случаев и, как полагают, передается по аутосомно-доминантному типу. [46] У пациентов с БАС было зарегистрировано множество различных мутаций SOD1 с разной степенью прогрессирования. Белок SOD1 отвечает за разрушение встречающихся в природе, но вредных супероксидных радикалов, вырабатываемых митохондриями . Большинство мутаций SOD1, связанных с БАС, представляют собой мутации усиления функции, при которых белок сохраняет свою ферментативную активность, но агрегируется в мотонейронах, вызывая токсичность. [47] [48] Нормальный белок СОД также участвует в других случаях БАС из-за потенциального клеточного стресса. [49] Была разработана модель БАС на мышах, основанная на мутациях SOD1, обеспечивающих усиление функции. [50]
- c9orf72
- Было обнаружено, что ген c9orf72 имеет гексануклеотидный повтор в некодирующей области гена, связанного с БАС и БАС-ЛВД. [51] Эти гексануклеотидные повторы могут присутствовать более чем в 40% случаев семейного БАС и в 10% спорадических случаев. C9orf72, вероятно, действует как фактор обмена гуанина для небольшой ГТФазы , но это, вероятно, не связано с основной причиной БАС. [52] Гексануклеотидные повторы, вероятно, вызывают клеточную токсичность после того, как они отделяются от транскриптов мРНК c9orf72 и накапливаются в ядрах пораженных клеток. [51]
- UBQLN2
- Ген UBQLN2 кодирует белок убиквилин 2, который отвечает за контроль деградации убиквитинированных белков в клетке. Мутации в UBQLN2 препятствуют деградации белка, что приводит к нейродегенерации за счет аномальной агрегации белка. [53] Эта форма БАС связана с Х-хромосомой и доминантно наследуется, а также может быть связана с деменцией .
Эпигенетическое лечение ингибиторами HDAC
[ редактировать ]Пациенты с БАС и модели на мышах демонстрируют общее гипоацетилирование гистонов, которое в конечном итоге может вызвать апоптоз клеток. [54] В экспериментах на мышах ингибиторы HDAC противодействуют этому гипоацетилированию, реактивируют аберрантно подавленные гены и противодействуют инициации апоптоза. [13] [55] Кроме того, известно, что ингибиторы HDAC предотвращают образование агрегатов белка SOD1 in vitro. [56]
- Фенилбутират натрия
- Лечение фенилбутиратом натрия на мышиной модели БАС с SOD1 показало улучшение двигательных качеств и координации, уменьшение нервной атрофии и потери нейронов, а также увеличение набора веса. [9] [10] Высвобождение проапоптотических факторов также было прекращено, как и общее увеличение ацетилирования гистонов. [55] Испытание на людях с использованием фенилбутурата у пациентов с БАС показало некоторое увеличение ацетилирования гистонов, но в исследовании не сообщалось, улучшались ли симптомы БАС при лечении. [11]
- Вальпроевый синдром
- В исследованиях на мышах вальпроевая кислота восстанавливала уровни ацетилирования гистонов, повышала уровни факторов, способствующих выживанию, а у мышей наблюдалось улучшение двигательных качеств. [57] Однако, хотя препарат и отсрочил возникновение БАС, он не увеличил продолжительность жизни и не предотвратил денервацию . [58] Испытания вальпроевой кислоты на людях у пациентов с БАС не улучшили выживаемость и не замедлили прогрессирование. [12]
- Трихостатин А
- Испытания трихостатина А на моделях БАС на мышах восстановили ацетилирование гистонов в спинальных нейронах, уменьшили демиелинизацию аксонов и увеличили выживаемость мышей. [13]
Спинальная мышечная атрофия (СМА)
[ редактировать ]
Спинальная мышечная атрофия (СМА) — аутосомно-рецессивное заболевание двигательных нейронов, вызванное мутациями гена SMN1 . [59] Симптомы сильно различаются в зависимости от подгруппы СМА и стадии заболевания. Общие симптомы включают общую мышечную слабость и плохой мышечный тонус, включая конечности и дыхательные мышцы, что приводит к трудностям при ходьбе, дыхании и кормлении. В зависимости от типа СМА заболевание может проявляться от младенчества до взрослого возраста. Поскольку белок SMN обычно способствует выживанию мотонейронов, мутации в SMN1 приводят к медленной дегенерации мотонейронов, что приводит к прогрессирующему общесистемному истощению мышц. В частности, со временем снижение уровня белка SMN приводит к постепенной гибели альфа- мотонейронов в передних рогах спинного и головного мозга. Мышцы зависят от связей с двигательными нейронами и центральной нервной системой, чтобы стимулировать поддержание мышц, и поэтому дегенерация двигательных нейронов и последующая денервация мышц приводят к потере мышечного контроля и мышечной атрофии. Часто в первую очередь поражаются мышцы нижних конечностей, затем – верхние конечности, а иногда и мышцы дыхания и жевания. В целом проксимальные мышцы всегда поражаются больше, чем дистальные.
Генетическая причина
[ редактировать ]Спинальная мышечная атрофия связана с генетическими мутациями в гене SMN1 (выживание двигательного нейрона 1). Белок SMN широко экспрессируется в нейронах и выполняет множество функций внутри нейронов, включая построение сплайсосом , транспорт аксонов мРНК, рост нейритов во время развития и формирование нервно-мышечных соединений . Потеря причинной функции при СМА в настоящее время неизвестна.
SMN1 расположен в теломерной области 5-й хромосомы человека , а также содержит SMN2 в центромерной области. SMN1 и SMN2 почти идентичны, за исключением одного нуклеотидного изменения в SMN2, приводящего к появлению альтернативного сайта сплайсинга, где интрон 6 встречается с экзоном 8. Это изменение одной пары оснований приводит только к 10–20% транскриптов SMN2, что приводит к образованию полностью функционального белка SMN и 80 –90% транскриптов приводят к усеченному белку, который быстро разрушается. У большинства пациентов со СМА имеется 2 или более копий гена SMN2, причем большее количество копий приводит к снижению тяжести заболевания. [60] У большинства пациентов со СМА наблюдаются либо точечные мутации , либо делеция экзона 7, что часто приводит к образованию белкового продукта, подобного укороченной и деградированной версии белка SMN2. У пациентов со СМА это небольшое количество функционального белкового продукта SMN2 позволяет некоторым нейронам выжить.
Эпигенетическое лечение посредством активации гена SMN2
[ редактировать ]Хотя СМА не вызывается эпигенетическим механизмом, терапевтические препараты, воздействующие на эпигенетические метки, могут принести пациентам со СМА некоторое облегчение, остановив или даже обратив вспять прогрессирование заболевания. Поскольку пациенты со СМА с более высоким числом копий гена SMN2 имеют менее серьезные симптомы, исследователи предсказали, что эпигенетические препараты, которые увеличивают экспрессию мРНК SMN2, будут увеличивать количество функционального белка SMN в нейронах, что приведет к уменьшению симптомов СМА. Ингибиторы гистондеацетилазы (HDAC) являются основными соединениями, которые, как было протестировано, повышают экспрессию мРНК SMN2. Ингибирование HDAC может привести к гиперацетилированию локусов гена SMN2, что теоретически приведет к увеличению экспрессии SMN2. [41] Многие из этих ингибиторов HDAC (HDACi) сначала тестируются на мышиных моделях СМА, созданных в результате различных мутаций в мышином гене SMN1. Если на мышах будет наблюдаться улучшение и препарат не вызовет большого количества побочных эффектов или токсичности, его можно будет использовать в клинических испытаниях на людях. Испытания на людях всех перечисленных ниже ингибиторов HDAC чрезвычайно разнообразны и часто зависят от конкретного подтипа СМА у пациента.
- Квизиностат (JNJ-26481585)
- Квизиностат эффективен в низких дозах, что приводит к некоторому улучшению нервно-мышечной функции на мышиной модели СМА, но выживаемость не увеличивается. [61] Испытания на людях не проводились.
- Бутират натрия
- Бутират натрия был первым ингибитором HDAC, протестированным на моделях мышей со СМА. Это продлило продолжительность жизни мышей со СМА на 35% и показало повышенный уровень белка SMN в тканях спинного мозга. [28] [29] Однако бутират натрия до сих пор не использовался в исследованиях на людях.
- Фенилбутират натрия
- Фенилбутират натрия увеличивает полноразмерные транскрипты мРНК SMN2 в культуре клеток, но для сохранения результатов необходимо повторять применение препарата. [28] Испытания на людях показывают неоднозначные результаты: одно исследование показало повышение уровня транскриптов SMA в крови и улучшение двигательной функции. [30] но более крупное исследование не показало никакого влияния на прогрессирование заболевания или двигательную функцию. [29]
- Вальпроевая кислота
- Вальпроевая кислота, добавленная к клеткам пациентов со СМА, увеличила уровни мРНК и белка SMN2, и что препарат напрямую активирует промотор SMN2. [31] [32] В модели на мышах со СМА вальпроевая кислота была добавлена в питьевую воду и восстановила плотность двигательных нейронов и увеличила количество двигательных нейронов в течение 8 месяцев. [33] Испытания на людях чрезвычайно разнообразны, демонстрируя повышение уровня SMN2 и увеличение мышечной силы в некоторых исследованиях и абсолютное отсутствие эффектов в других исследованиях. [35] [34] [36] [37]
- М344
- M344 представляет собой бензамид, который показывает многообещающие результаты в культуре клеток фибробластов и повышает уровень факторов сплайсинга, которые, как известно, модулируют транскрипты SMN2, но препарат был признан токсичным, и исследования не дошли до испытаний in vivo. [27]
- Трихостатин А
- Лечение трихостатином А показывает многообещающие результаты на мышах. В одном исследовании трихостатин А в сочетании с дополнительным питанием на моделях СМА у мышей с ранним началом привел к улучшению двигательной функции и выживаемости, а также задержал прогрессирующую денервацию мышц. [38] Второе исследование на модели мышей со СМА показало увеличение транскриптов SMN2 при ежедневных инъекциях. [39] Испытания на людях не проводились.
- Вориностат (SAHA)
- Вориностат — ингибитор второго поколения, который достаточно нетоксичен и эффективен в культуре клеток при низких концентрациях. [40] и увеличивает ацетилирование гистонов на промоторе SMN2. [41] В модели на мышах со СМА лечение SAHA привело к увеличению веса, повышению уровня транскриптов SMN2 в мышцах и спинном мозге, а также к остановке потери двигательных нейронов и денервации. [42] Испытания на людях не проводились.
Миастения гравис
[ редактировать ]Миастения гравис — аутоиммунное заболевание, поражающее синапсы в нервно-мышечных соединениях, при котором антитела, вырабатываемые преимущественно в тимусе В-клетками, связываются с постсинаптическим никотиновым ацетилхолиновым рецептором (AChR), а также с другими постсинаптическими рецепторами NMJ (MuSK-R и низкочастотными рецепторами ацетилхолина). рецептор липопротеина плотности). Эти антитела включают антитела к рецептору ацетилхолина, антитела MuSK и антитела к белку 4, родственному рецептору липопротеина низкой плотности (LRP4-Ab). [62] Связывание антител с соответствующими рецепторами вызывает разрушение этих рецепторов, что приводит к уменьшению количества постсинаптических ацетилхолинергических рецепторов и снижению общего транспорта ацетилхолина. Симптомы заболевания включают мышечную слабость, которая утомляется из-за чрезмерной нагрузки, но проходит после отдыха. Отличительные симптомы мышечной слабости включают птоз, двоение в глазах, дисфагию, а также аномальную речь. [63]
Миастения гравис — относительно редкое заболевание, встречающееся примерно у 3–30 человек на 100 000, но за последние пару десятилетий ее число возросло. Существует два варианта миастении гравис в зависимости от возраста и пола: миастения с ранним началом, которая чаще встречается у женщин, и миастения с поздним началом, которая чаще встречается у мужчин. [63]
Эпигенетические факторы
[ редактировать ]Было проведено обширное исследование генетической основы миастении, однако данные не свидетельствуют о том, что это наследственное заболевание. [64] Также были проведены обширные исследования эпигенетического вклада в развитие миастении. [65] Метилирование ДНК и некодирующие РНК, такие как микроРНК (микроРНК) и длинные некодирующие РНК (днРНК), являются эпигенетическими факторами, которые играют значительную роль в повышении вероятности приобретения миастении гравис. Кроме того, тимус является ключевым органом иммунного ответа, на который часто негативно влияют аномальная экспрессия микроРНК и метилирование ДНК.
микроРНК
[ редактировать ]МикроРНК (миРНК) представляют собой одноцепочечные некодирующие РНК, которые связывают мРНК-мишени. Отсюда они могут регулировать экспрессию генов, ингибируя трансляцию или разрушая цепь мРНК, часто в В-клетках и Т-клетках иммунологического процесса. Что касается миастении гравис, аномальная функция микроРНК связана с иммунорегуляторным патогенезом, и каждая микроРНК имеет свои собственные уникальные последующие эффекты.
Тимус – важный эндокринный орган, участвующий в развитии миастении. При нормальном, здоровом развитии тимус со временем уменьшается в размерах. У пациентов с миастенией, связанной с тимусом, наблюдаются корреляции с тимомами при миастении с поздним началом, а также с гиперплазией тимуса с зародышевыми центрами при миастении с ранним началом, и каждое из этих состояний можно частично объяснить нерегулярной функцией микроРНК. [66] У субъектов с поздней миастенией было показано, что экспрессия микроРНК-12a-5p увеличивается при миастении, связанной с тимомой. МиРНК-12a-5p ингибирует экспрессию гена FoxP3, гена, который, как известно, связан с нормальным развитием тимуса и изменения которого связывают с тимомами. [67] Кроме того, была обнаружена связь между миастенией, ассоциированной с тимомой, и снижением экспрессии миР-376a/миР-376c. Известно, что аутоиммунная регуляция снижается при миастении, связанной с тимомой, а в клетках тимуса с пониженной аутоиммунной регуляцией наблюдается одновременное снижение экспрессии миР-376a, миР-376c и миРНК-12a-5p. [67] У пациентов с миастенией с ранним началом 61 микроРНК было обнаружено либо значительно сниженное, либо повышенное регулирование. Было обнаружено, что наиболее подавленной миРНК оказалась миР-7-5p, геном-мишенью которой является CCL21. Известно, что CCL21 аберрантно рекрутирует B-клетки в тимусе у пациентов с миастенией с ранним началом, и было обнаружено, что он высоко экспрессируется у пациентов с миастенией с ранним началом, что потенциально объясняет аномально большое количество B-клеток, обнаруживаемых при гиперплазии тимуса. [68]
Помимо микроРНК, соответствующих измененной функции тимуса, существует несколько других ключевых микроРНК, которые коррелируют с миастенией гравис. Было показано, что кластер МиР-15 (миР-15а, миР-15b и миР-15c) связан с аутоиммунитетом, поскольку его подавление увеличивает экспрессию CXCL10, гена-мишени, участвующего в передаче сигналов Т-клеток. Также было показано, что экспрессия CXCL10 увеличивается в тимусе пациентов с миастенией гравис. [69] Кроме того, было обнаружено повышение уровня экспрессии миР-146 у пациентов с миастенией гравис. У этих пациентов с повышенным уровнем экспрессии миР-146 наблюдалось одновременное увеличение количества белков, которые соответствуют широкому спектру иммунных ответов, в частности TLR4, CD40 и CD80. [70]
Метилирование ДНК
[ редактировать ]Метилирование ДНК — это эпигенетический процесс, при котором метильные группы добавляются либо к адениновым, либо к цитозиновым основаниям, что приводит к репрессии этой последовательности при возникновении метилирования цитозина. [71] Было обнаружено, что метилирование ДНК является фактором повышения вероятности приобретения миастении, хотя эта тема не была широко исследована. Исследования в Китае выявили, что ген CTLA-4 (антиген-4 цитотоксических Т-лимфоцитов) сильно метилирован у пациентов с миастенией по сравнению с контрольными группами на протяжении всего периода заболевания. Ген CTLA-4 продуцирует одноименный антиген, который представлен на Т-клетках-киллерах и позволяет подавлять иммунный ответ. Метилирование этого гена подавляет выработку антигена CTLA-4 — закономерность, наблюдаемая у значительного большинства пациентов с миастенией — и может объяснить повышенный иммунный ответ, наблюдаемый при миастении. [72] Кроме того, у больных миастенией с аномалиями тимуса (примерно 10–20% всех больных миастенией) [73] имели даже более высокий уровень метилирования CTLA-4, чем другие пациенты с миастенией. Причины гиперметилирования определенных генов в этих случаях не изучены, но информация о миастении в основном указывает на усиление регуляции генов ДНК-метилтрансферазы DNMT1, DNMT3A и DNMT3B у пациентов с миастенией gravis. [74]
Помимо метилирования CTLA-4, у пациентов с миастенией с поздним началом, ассоциированной с тимомой, наблюдалось гиперметилирование гена рецептора, усиливающего секрецию гормона роста. [75] Гиперметилирование рецепторов, стимулирующих секрецию гормона роста, обнаруживается при широком спектре раковых заболеваний, однако только недавно оно было коррелировано с развитием миастении, связанной с тимомой. Хотя он наблюдается примерно у 1/4 пациентов с тимомой, связанной с миастенией гравис, он не является надежным биомаркером заболевания, и его влияние на прогрессирование заболевания недостаточно изучено.
Длинная нкРНК
[ редактировать ]Длинные нкРНК (днРНК) представляют собой второй тип некодирующих РНК, которые являются ключевыми посттранскрипционными модификаторами экспрессии генов, кодирующих белок. Они также играют значительную роль при миастении. Их аберрантная регуляция может вызывать дифференциальную экспрессию в нижестоящих генах. Например, дифференциальная экспрессия днРНК гамма-антсмысловой РНК интерферона отрицательно регулирует экспрессию HLA-DRB и HLA-DOB. [75] два гена участвуют в аутоиммунном ответе организма путем дифференциации эндогенных и чужеродных белков. [76] Как наблюдалось у пациентов с миастенией с пониженной регуляцией летальной днРНК (let)-7, также было обнаружено, что уровень днРНК let-7 отрицательно коррелировал с уровнями интерлейкина (IL)-10, гена, ответственного за ингибирование секреции/активации цитокинов. презентация антигена и активность макрофагов, [77] но и для проявления противоопухолевого эффекта. [78] Таким образом, отрицательная корреляция между уровнями lncRNA let-7 и IL-10 и его специфическим влиянием на развитие миастении неоднозначна.
Помимо аберрантной регуляции нижестоящих генов-мишеней, lncRNA также влияет на экспрессию, действуя как конкурирующая эндогенная РНК (ceRNA). Теория конкурирующих эндогенных РНК утверждает, что транскрипты, имеющие общие сайты связывания микроРНК, могут конкурировать за связывание этих идентичных микроРНК, и таким образом днРНК могут связывать микроРНК, регулируя их нижестоящую связывающую активность и влияя на их функцию. В случае миастении ген хозяина днРНК малой ядрышковой РНК (SNHG) 16 регулирует экспрессию IL-10 путем адсорбции let-7c-5p, микроРНК, которая обычно связывается с IL-10, в качестве конкурирующей эндогенной РНК.
Эпигенетические методы лечения
[ редактировать ]Диагностика миастении гравис, индивидуальный прогноз и уровень необходимого лечения могут быть определены путем определения количества циркулирующей микроРНК.
Иммунодепрессанты представляют собой большую категорию в клинических исследованиях лечения миастении гравис, поскольку они уменьшают гиперактивный иммунологический ответ в Т-клетках, презентирующих антигены, связывающие рецептор ацетилхолина. [66] Исследования показывают, что благодаря сверхэкспрессии миР-146 пациенты с ранней миастенией гравис могут оказывать антигенспецифическое супрессивное действие. Это имеет значение для снижения иммунного ответа у пациентов с миастенией и улучшения прогноза. Аналогично, доказано, что миР-155 коррелирует с воспалением тимуса, связанным с миастенией гравис, и иммунным ответом. Проводятся исследования, в которых репрессия миР-155 может уменьшить эти аберрантные эффекты. [66] Наконец, постоянно показано, что уровень миР-150-5p и миР-21-5p микроРНК повышен у пациентов с миастенией гравис с антителами к ацетилхолинергическим рецепторам (в отличие от MuSK-связывающего варианта миастении гравис), поэтому эти две микроРНК являются надежными биомаркерами. в выявлении этого варианта миастении. [79]
Нейродегенеративные заболевания центральной нервной системы
[ редактировать ]Болезнь Альцгеймера (БА)
[ редактировать ]Болезнь Альцгеймера (БА) является наиболее распространенной формой деменции среди пожилых людей. Заболевание характеризуется поведенчески хроническим и прогрессирующим снижением когнитивных функций, начиная с кратковременной потери памяти, а неврологически – накоплением неправильно свернутого тау-белка и связанными с ним нейрофибриллярными клубками , а также бета-амилоидными сенильными бляшками . Было идентифицировано несколько генетических факторов, способствующих развитию БА, включая мутации белка-предшественника амилоида ( АРР ) и генов пресенилинов 1 и 2 , а также семейное наследование аллели аполипопротеина Е эпсилон 4. В дополнение к этим общим факторам существует ряд другие гены, экспрессия которых изменена при болезни Альцгеймера, некоторые из которых связаны с эпигенетическими факторами.
Эпигенетические факторы
[ редактировать ]
- нкРНК
- нкРНК, которая кодируется антисмысловой интроном гена фермента, расщепляющего бета-амилоид, BACE1, участвует в развитии болезни Альцгеймера. [6] Эта нкРНК, BACE1-AS (антисмысловая), которая перекрывает экзон 6 BACE1 , участвует в повышении стабильности транскрипта мРНК BACE1 . Как следует из названия этого гена, BACE1 представляет собой ферментативный белок, который расщепляет белок-предшественник амилоида до нерастворимой бета-формы амилоида, которая затем агрегируется в старческие бляшки. Благодаря повышенной стабильности мРНК BACE1 в результате BACE1-AS больше мРНК BACE1 доступно для трансляции в белок BACE1.
- микроРНК
- Не было доказано, что факторы играют роль в прогрессировании БА. микроРНК участвуют в посттранскрипционном молчании генов посредством ингибирования трансляции или участия в путях РНКи . Некоторые исследования показали усиление экспрессии микроРНК-146a, которая по-разному регулирует нейроиммунную экспрессию киназ IRAK1 и IRAK2, связанных с нейроиммунным интерлейкином-1R, в мозге человека с AD, в то время как другие исследования показали повышение или понижение уровня экспрессии микроРНК-9 в головном мозге. [80]
- Метилирование ДНК
- В случаях болезни Альцгеймера наблюдалось глобальное гипометилирование ДНК и геноспецифическое гиперметилирование, хотя результаты в разных исследованиях различались, особенно в исследованиях человеческого мозга. Гипотетически, глобальное гипометилирование должно быть связано с глобальным увеличением транскрипции, поскольку CpG-островки наиболее распространены в промоторах генов; однако ген-специфическое гиперметилирование может указывать на то, что эти гиперметилированные гены подавляются метками метилирования. Обычно репрессивное гиперметилирование генов, связанных с обучением и памятью, наблюдается в сочетании с дерепрессивным гипометилированием нейровоспалительных генов и генов, связанных с патологическим проявлением болезни Альцгеймера. Снижение метилирования было обнаружено в нейронах височной коры головного мозга, связанных с долговременной памятью, у монозиготных близнецов с болезнью Альцгеймера по сравнению со здоровыми близнецами. [81] Глобальное гипометилирование динуклеотидов CpG также наблюдалось в гиппокампе. [82] и во II слое энторинальной коры [83] человеческих пациентов с AD, оба из которых восприимчивы к патологии AD. Эти результаты, полученные с помощью иммуноанализа, были оспорены исследованиями, которые исследовали последовательность ДНК с помощью бисульфитного секвенирования , метода трансформации CpG, который чувствителен к статусу метилирования CpG, в котором наблюдалось глобальное гипометилирование. [84] [85]
- ЦОГ-2
- На уровне отдельных генов происходит гипометилирование и, следовательно, дерепрессия ЦОГ-2 , ингибирование которого уменьшает воспаление и боль, а также гиперметилирование BDNF — нейротрофического фактора, важного для долговременной памяти. [85] экспрессия CREB , зависимого от активности фактора транскрипции, участвующего в регуляции BDNF Также было показано, что среди многих других генов, также гиперметилирована и, таким образом, подавляется в мозге с болезнью Альцгеймера, что еще больше снижает транскрипцию BDNF . [85] Кроме того, было показано, что синаптофизин ( SYP ) , основной ген, кодирующий белок синаптических пузырьков, гиперметилирован и, следовательно, репрессирован, а фактор транскрипции NF-κB , который участвует в передаче иммунных сигналов, гипометилирован и, следовательно, депрессирован. [85] В совокупности эти результаты пролили свет на роль нарушения регуляции генов, участвующих в обучении, памяти и синаптической передаче, а также в иммунном ответе.
- Гипометилирование
- наблюдался в промоторах пресенилина 1 , [86] GSK3beta , фосфорилирующий тау-белок, [87] и БАСЕ1 , [88] фермент, расщепляющий АРР в бета-форму амилоида, которая, в свою очередь, агрегируется в нерастворимые старческие бляшки. Репрессивное гиперметилирование, вызванное бета-амилоидом, наблюдалось в промоторе NEP , гена неприлизина, который является основным ферментом, очищающим бета-амилоид в мозге. [89] Это подавление НЭП может привести к накоплению старческих бляшек; в сочетании с наблюдаемым увеличением BACE1-AS в мозге при AD и соответствующим увеличением белка BACE1 и бета-амилоида, [6] Несколько уровней эпигенетической регуляции могут участвовать в контроле образования, клиренса или агрегации бета-амилоида и отложения старческих бляшек. Возраст может иметь некоторое влияние на уровни метилирования ДНК на определенных промоторах генов, поскольку одно исследование обнаружило более высокие уровни метилирования промоторов APP у пациентов с AD до 70 лет, но более низкие уровни метилирования у пациентов старше 70 лет. [90] Исследования дифференциального метилирования ДНК в мозге человека с болезнью Альцгеймера остаются в значительной степени безрезультатными, возможно, из-за высокой степени изменчивости между людьми и многочисленных комбинаций факторов, которые могут привести к болезни Альцгеймера.
- Гистоновые метки
- Ацетилирование остатков лизина на хвостах гистонов обычно связано с активацией транскрипции, тогда как деацетилирование связано с репрессией транскрипции. Существует мало исследований, исследующих специфические метки гистонов при БА. Эти исследования выявили снижение ацетилирования лизинов 18 и 23 на N-концевых хвостах гистона 3 (H3K18 и H3K23 соответственно). [91] и увеличение HDAC2 в мозге при болезни Альцгеймера [92] - обе отметки связаны с репрессией транскрипции. Возрастное снижение когнитивных функций было связано с нарушением регуляции ацетилирования H4K12, когнитивного эффекта, который был восстановлен у мышей путем индукции этой метки. [93]
Лечение
[ редактировать ]Лечение для профилактики или лечения болезни Альцгеймера оказалось проблематичным, поскольку болезнь является хронической и прогрессирующей, а многие эпигенетические препараты действуют глобально, а не генно-специфическим образом. Как и другие потенциальные методы лечения для предотвращения или облегчения симптомов БА, эти методы лечения не излечивают, а лишь временно облегчают симптомы заболевания, подчеркивая хронический, прогрессирующий характер АД и изменчивость метилирования в мозге при БА.
- Фолиевая кислота и другие витамины группы B.
- Витамины группы B участвуют в метаболическом пути, который приводит к выработке SAM. SAM является донором метильной группы, используемой ДНК-метилтрансферазами (DNMT) для метилирования CpG. Используя модели животных, Fuso et al. продемонстрировали восстановление метилирования ранее гипометилированных промоторов пресенилина 1 , BACE1 и APP. [94] – гипотетически стабильная эпигенетическая модификация, которая должна подавлять эти гены и замедлять прогрессирование болезни Альцгеймера. Также было показано, что диетические добавки SAM уменьшают окислительный стресс и задерживают накопление неврологических признаков AD, таких как бета-амилоид и фосфорилированный тау-белок, у трансгенных мышей с AD.
- НЕ
- Хан и его коллеги продемонстрировали потенциальную роль нейроглобинина , связанной с амилоидом в ослаблении нейротоксичности . [95] 5-аза-2'-дезоксицитидин (АЗА или децитабин), ингибитор DNMT, продемонстрировал некоторые доказательства регуляции экспрессии нейроглобина, хотя это открытие не было проверено на моделях AD. [96]
- Гистон-направленное лечение
- Хотя исследования меток гистонов в мозге при болезни Альцгеймера немногочисленны, в нескольких исследованиях изучалось влияние HDACis на лечение болезни Альцгеймера. Ингибиторы HDAC I и II классов, такие как трихостатин А, вориностат и бутират натрия, а также HDAC класса III, такие как никотинамид, оказались эффективными при лечении симптомов на животных моделях AD. Несмотря на то, что HDACis является многообещающим терапевтическим средством на животных моделях, исследования долгосрочной эффективности HDACis и испытания на людях еще не проводились.
- Бутират натрия
- Бутират натрия относится к HDACi класса I и II и, как было показано, восстанавливает обучаемость и память через 4 недели. [14] уменьшать фосфорилированный тау-белок и восстанавливать плотность дендритных шипов в гиппокампе трансгенных мышей с AD. [15] Ацетилирование гистонов в результате диффузного применения бутирата натрия особенно распространено в гиппокампе, а гены, участвующие в обучении и памяти, показали повышенное ацетилирование у мышей с AD, получавших этот препарат. [16]
- Трихостатин А
- Трихостатин А также является HDACi класса I и II, который спасает обучение страху в парадигме обусловливания страха у трансгенных мышей с AD до уровней дикого типа посредством ацетилирования лизиновых хвостов гистона 4. [18]
- Вориностат
- Вориностат представляет собой HDACi класса I и II, который, как было показано, особенно эффективен для ингибирования HDAC2 и восстановления функций памяти в моделях дефицита обучения, не связанных с AD. [97] Одно исследование показало, что вориностат эффективен в устранении дефицита контекстуальной памяти у трансгенных мышей с болезнью Альцгеймера. [17]
Хантингтон (HD)
[ редактировать ]
Болезнь Хантингтона (БГ) — наследственное заболевание, вызывающее прогрессирующую дегенерацию нейронов коры головного мозга и полосатого тела головного мозга. [98] что приводит к потере двигательных функций (непроизвольным сокращениям мышц), снижению когнитивных способностей (что в конечном итоге приводит к слабоумию) и изменениям в поведении. [7]
Генетика и основные причины
[ редактировать ]Болезнь Хантингтона вызвана аутосомно-доминантной мутацией, увеличивающей количество повторов кодона глутамина (CAG) в гене Хантингтина (Htt). [98] Ген Htt кодирует белок хантингтин, который играет роль в нормальном развитии, но его точная функция остается неизвестной. [99] Длина этого повтора CAG коррелирует с возрастом начала заболевания. У среднестатистического человека без болезни Хантингтона в гене Htt присутствует менее 36 CAG-повторов. Когда эта длина повтора превышает 36, начало деградации нейронов и физические симптомы болезни Хантингтона могут варьироваться от 5 лет (повтор CAG > 70) до 80 лет (повтор CAG < 39). [100]
Это расширение CAG приводит к подавлению мРНК определенных генов, снижению ацетилирования гистонов и увеличению метилирования гистонов. [101] [102] Точный механизм того, как этот повтор вызывает нарушение регуляции генов, неизвестен, но модификация эпигенома может сыграть свою роль. При болезни Хантингтона с ранним началом (в возрасте 5–15 лет) как у трансгенных мышей, так и у мышей в полосатых клеточных линиях наблюдается специфическое для мозга гипоацетилирование гистона H3 и снижение ассоциации гистонов со специфическими подавленными генами в полосатом теле (а именно, Bdnf, Cnr1, Drd2 – рецептор дофамина 2 и Penk1 – препроэнкефалин). [103] Как при позднем, так и при раннем начале болезни Хантингтона коровые гистоны H3 и H4, связанные с этими генами с пониженной регуляцией у мутантов Htt, имеют гипоацетилирование (снижение ацетилирования) по сравнению с Htt дикого типа. [102] [103] Этого гипоацетилирования достаточно, чтобы вызвать более плотную упаковку хроматина и подавление мРНК. [102]
Наряду с гипоацетилированием H3, как у людей, так и у мышей с мутантным Htt наблюдаются повышенные уровни триметилирования лизина-9 гистона H3. [101] Это увеличение триметилирования H3-K9 связано с повышенной экспрессией метилтрансферазы ESET/SETDB1 (ERG-ассоциированный белок с доменом SET (ESET)), которая нацеливается на остатки H3-K9 и триметилирует их. [101] Предполагается, что это гиперметилирование может быть причиной возникновения специфической репрессии генов у мутантов Htt. [101]
Ингибиторы HDAC
[ редактировать ]У пациентов Хантингтона, а также на моделях мышей и дрозофилы наблюдается гипоацетилирование гистонов H3 и H4. В настоящее время не существует методов лечения этого заболевания, но были протестированы многочисленные ингибиторы HDAC, и было показано, что они обращают вспять определенные симптомы, вызванные мутацией Htt.
- Бутират натрия
- Лечение бутиратом натрия замедляло дегенерацию нейронов на моделях дрозофилы. [19] Обработка бутиратом натрия также увеличивала ацетилирование гистона H3 и нормализовала уровни мРНК для мутантных генов с пониженной регуляцией Htt. [103]
- Вальпроевая кислота
- Обработка вальпроевой кислотой повышала уровни ацетилирования мутантных Htt H3 и H4, сравнимые с Htt дикого типа на моделях дрозофилы. [19]
- Фенилбутират натрия
- Испытания II фазы фенилбутирата натрия на людях с приемом от 12 до 15 г/день показали восстановление уровней мРНК репрессированных мутантных генов Htt, но также имели неблагоприятные побочные эффекты, такие как тошнота, головные боли и нестабильность роста. [104] Также было показано, что фенилбутират увеличивает ацетилирование гистонов, снижает метилирование гистонов, увеличивает выживаемость и снижает скорость деградации нейронов на моделях мышей с мутацией Htt. [20]
- Трихостатин А
- Лечение трихостатином А (TSA) повышало уровни ацетилирования мутантных Htt H3 и H4, сравнимые с Htt дикого типа на моделях дрозофилы. [19] Также было показано, что лечение TSA увеличивает ацетилирование лизина 40 альфа-тубулина в клетках полосатого тела мышей и увеличивает внутриклеточный транспорт BDNF, нейротрофического фактора головного мозга, который участвует в росте и поддержании нервов в мозге. [105] [21]
- Вориностат (SAHA)
- Лечение вориностатом замедляло дегенерацию фоторецепторов и увеличивало продолжительность жизни взрослых дрозофил, мутантных по Htt. [19] Как и TSA, обработка SAHA увеличивала ацетилирование лизина 40 альфа-тубулина в клетках полосатого тела мышей, а также увеличивала внутриклеточный транспорт BDNF.
Болезнь Паркинсона (БП)
[ редактировать ]
Болезнь Паркинсона (БП) характеризуется прогрессирующей дегенерацией дофаминергических нейронов черной субстанции по неизвестным причинам. Некоторые гены и факторы окружающей среды (например, воздействие пестицидов) могут играть роль в возникновении БП. Отличительные признаки включают мутации гена альфа-синуклеина, SNCA , а также генов PARK2 , PINK1 , UCHL1 , DJ1 и LRRK2 , а также фибриллярное накопление телец Леви из неправильно свернутого альфа-синуклеина. Симптомы наиболее заметно проявляются в нарушениях движений, включая тряску, ригидность, нарушение контролируемых движений, медленную и затрудненную ходьбу. Поздние стадии заболевания приводят к слабоумию и депрессии. Леводопа и дофаминергическая терапия могут облегчить симптомы, хотя лечения, способного остановить прогрессирование заболевания, не существует.
Эпигенетические факторы
[ редактировать ]- нкРНК
- Снижение уровня миР-133b коррелировало с уменьшением количества дофаминергических нейронов в среднем мозге пациентов с БП. [106] Между тем, миР-132 отрицательно коррелирует с дифференцировкой дофаминергических нейронов в среднем мозге. [107] миР-7 и миР-153 снижают уровни альфа-синуклеина (характерный признак БП), но их уровень снижается в головном мозге с БП. [108]
- Метилирование ДНК
- В нейронах больных БП наблюдается гипометилирование последовательности, кодирующей фактор некроза опухоли (TNF-альфа) , сверхэкспрессия которой приводит к апоптозу нейронов. [109] В спинномозговой жидкости пациентов с БП также наблюдается повышенный уровень ФНО-альфа. [110] Исследования показывают, что может существовать связь между метилированием ДНК и экспрессией SNCA. [111] [112] Кроме того, модели на людях и мышах показали снижение уровней ядерного DNMT1 у субъектов с БП, что приводит к гипометилированным состояниям, связанным с репрессией транскрипции. [113]
- Гистоновые метки
- альфа-синуклеин, белок, кодируемый SNCA , может связываться с гистонами и предотвращать их ацетилирование совместно с HDAC HDAC1 и Sirt2. [26] [114] Кроме того, было продемонстрировано, что альфа-синуклеин связывает гистон 3 и ингибирует его ацетилирование у дрозофилы . [26] Истощение дофамина при болезни Паркинсона связано с репрессивными модификациями гистонов, включая снижение H3K4me3 и более низкие уровни ацетилирования лизина H3 и H4 после терапии леводопой (распространенное лечение БП).
Лечение
[ редактировать ]Эпигенетических методов лечения, протестированных на моделях БП, немного, хотя были проведены некоторые многообещающие исследования. Большинство исследованных на данный момент методов лечения направлены на модификации гистонов и анализ их роли в обеспечении экспрессии и активности альфа-синуклеина. Пестициды и паракват усиливают ацетилирование гистонов, вызывая нейротоксические эффекты, аналогичные тем, которые наблюдаются при БП, такие как апоптоз дофаминергических клеток. [115] Несмотря на это, лечение HDACis [116] по-видимому, оказывает нейропротекторное действие.
- Бутират натрия
- Несколько исследований с использованием различных моделей животных показали, что бутират натрия может быть эффективен в снижении нейротоксичности, связанной с альфа-синуклеином. [22] [23] У дрозофилы бутират натрия улучшил двигательные нарушения и снизил уровень ранней смертности. [24]
- Вальпроевая кислота
- В индуцибельной крысиной модели БП вальпроевая кислота оказывала нейропротекторное действие, предотвращая транслокацию альфа-синуклеина в ядра клеток. [25]
- Вориностат
- В модели БП у дрозофилы со сверхэкспрессией альфа-синуклеина вориностат (а также бутират натрия) снижал нейротоксичность, опосредованную альфа-синуклеином. [26]
- ингибирование siRNA SIRT2
- Лечение SIRT2, ингибирующим миРНК, приводит к снижению нейротоксичности альфа-синуклеина AK-1 или AGK-2. [114]
Рассеянный склероз
[ редактировать ]Рассеянный склероз (РС) — демиелинизирующее нейродегенеративное заболевание, причина которого не имеет подтвержденной причины, но широко считается по своей природе аутоиммунным заболеванием. [117] На это указывает демиелинизация нервов головного и спинного мозга. Его симптомы уникальны по своей природе и различаются, но включают те, которые имеют дегенеративные последствия в глазах и конечностях. Они могут проявляться в виде онемения или атрофии, ощущений шока, паралича, а также отсутствия координации или тремора в конечностях. Внутри глаза рассеянный склероз может вызвать нечеткость зрения, двоение в глазах, боль или потерю зрения. Эффекты рассеянного склероза могут проявляться и в других сферах организма, но в основном характеризуются этими основными симптомами. Некоторые из них могут включать потерю сексуальной или выделительной функции и эпилепсию. Хотя существует несколько подкатегорий рассеянного склероза, в большинстве случаев заболевание имеет рецидивирующий характер, когда рецидивы симптомов могут не возникать в течение длительных периодов времени, что еще больше увеличивает неопределенность заболевания. Не существует известного лечения рассеянного склероза, но после рецидива можно принять меры для восстановления потери функций, а симптомы можно смягчить с помощью терапевтических или медицинских средств. [118]
Эпигенетические факторы
[ редактировать ]Из-за внешних факторов, которые предшествуют рассеянному склерозу, и наследственности, обычно возникающей у матери, считается, что заболевание имеет эпигенетическую причину. Некоторыми факторами, которые могут увеличить заболеваемость рассеянным склерозом, являются курение, дефицит витаминов и некоторые вирусные инфекции в анамнезе, которые могут вызывать эпигенетические изменения. [119]
Аллель человеческого лейкоцитарного антигена DRB1*15
[ редактировать ]Гаплотип человеческого лейкоцитарного антигена-DRB1*15 является потенциальным фактором риска развития рассеянного склероза. Из-за повышенной вероятности передачи аллеля человеческого лейкоцитарного антигена-DRB1*15 матери их детям, это способствует более распространенному распространению рассеянного склероза у матери. Считается, что HLA-DRB1 регулируется эпигенетическими средствами. Предполагается, что корреляция MS и этого аллеля обусловлена наличием гипометилирования в CpG-островке HLA-DRB1, и те, кто несет этот аллель, имеют тенденцию проявлять это гипометилирование. Экзон 2 HLA-DRB1 представляет собой особую область, где данные показали, что метилирование играет важную роль в регуляции. Исследования предоставили доказательства того, что изменение HLA-DRB1 DMR, который представляет собой механизм, регулируемый метилированием, который, в свою очередь, регулирует повышенную экспрессию HLA-DRB1, демонстрирует повышенный риск развития рассеянного склероза и проявления заболевания. [119] [120]
микроРНК
[ редактировать ]Более высокие уровни экспрессии определенных типов микроРНК часто наблюдаются в мозгу больных, что указывает на связь этих типов микроРНК и рассеянного склероза. Более высокая экспрессия миР-155 и миР-326 часто связана с дифференцировкой CD4+Т-клеток, и при этой дифференцировке возникают случаи аутоиммунного энцефалита, что является связью, с которой считается, что курение может вызывать эпигенетические изменения, которые повышают восприимчивость к РС. Более высокие уровни экспрессии миР-18b, миР-493, миР-599 и миР-96 часто наблюдаются у пациентов с диагнозом РС. Обнаружение миР-145, по-видимому, является многообещающим будущим диагностическим инструментом благодаря его высокой специфичности 90% и чувствительности 89,5% при анализе цельной крови из-за его способности отличать здоровых пациентов от пациентов с рассеянным склерозом. Симптомом, связанным с пациентами с рассеянным склерозом, являются поражения белого вещества головного мозга, и эти поражения при биопсии показали более высокую экспрессию миР-155, миР-326 и миР-34а. Это особенно примечательно в связи с тем, что сверхэкспрессия этих микроРНК вызывает подавление CD47, что приводит к фагоцитозу миелина из-за роли CD47 в ингибировании активности макрофагов. [121]
Метилирование ДНК
[ редактировать ]Пациентов с рассеянным склерозом можно идентифицировать путем наблюдения за аномальными паттернами метилирования ДНК в генах, ответственных за воспаление и экспрессию факторов миелинизации. Метилирование происходит в геномной области, CpG-островке, и необходимо для регуляции транскрипции. Метилированная область CpG обычно является механизмом, который подавляет ген, тогда как гипометилированная область способна индуцировать транскрипцию. Было показано, что с использованием ингибиторов метилирования можно добиться более высокой пролиферации Т-клеток за счет предотвращения молчания. Нарушения в паттерне метилирования увеличивают выработку аутореактивных CD4+T. Гипометилирование областей CpG гена PAD2, регулятора MBP, который, в свою очередь, регулирует миелин, также связано с более частыми случаями рассеянного склероза. Это гипометилирование приводит к сверхэкспрессии гена PAD2. Эти закономерности наблюдались в белом веществе пациентов с рассеянным склерозом. Хотя метилирование является индикатором рассеянного склероза, его эффекты более специфичны для локализации, например, при рассеянном склерозе, где эти закономерности наблюдаются в белом веществе. [121]
Модификации гистонов
[ редактировать ]Ассоциацию аномальной модификации гистонов у пациентов с рассеянным склерозом можно обнаружить при поражениях, расположенных в головном мозге, причем большинство случаев этого наблюдается у пациентов с течением времени и при поражениях, расположенных в лобной доле. Более высокий уровень ацетилирования гистонов можно наблюдать у пациентов, страдающих с течением времени, но этому противодействуют более низкие случаи ацетилирования гистонов в поражениях, обнаруженных в мозге на ранних стадиях заболевания. Механизмы, с помощью которых модификации гистонов влияют на прогрессирование рассеянного склероза, не подтверждены, но изменения ацетилирования часто связаны с заболеванием.
Лечение
[ редактировать ]Ингибиторы HDAC
[ редактировать ]Трихостатин
Положительные реакции наблюдались в исследованиях на животных с использованием этого ингибитора HDAC, что связано с опосредованием воспалительных путей и, таким образом, приводит к снижению случаев воспалительных реакций в головном мозге. Также было показано, что он эффективен в снижении уровня инвалидности, когда мыши находились на стадии рецидива рассеянного склероза. Влияние трихостатина на симптомы недостаточно хорошо известно, но считается, что он способствует увеличению ацетилирования гистонов H3 и H4 в CD4+T-клетках, где у пациентов с рассеянным склерозом часто наблюдаются различия в уровнях ацетилирования этих гистонов, чего у контрольных пациентов нет.
Вориностат
Испытания на животных проводились наряду с тестированием миелоидных дендритных клеток человека. О механизмах действия вориностата известно немного; однако наблюдалась регуляция экспрессии цитокинов Th1/Th17, которые отвечают за индуцирование воспаления, тем самым уменьшая случаи воспаления и демиелинизации. Также наблюдалось снижение пролиферации Т-клеток, аналогично тому, как трихостатин опосредует симптомы заболевания. [122]
Вальпроповая кислота
В испытаниях на животных было показано, что вальпроповая кислота дает положительные результаты в смягчении заболевания путем регулирования тяжести и продолжительности рассеянного склероза. Его механизм заключается в уменьшении презентации микроРНК. Его механизм наблюдался у крыс путем смещения Th1 и Th17 на Th2 (ответственный за индуцирование воспаления), тем самым подавляя экспрессию микроРНК в воспалительных цитокинах, механизмах, опосредующих опухоль, и позвоночнике. Это еще один случай, когда присутствует регуляция экспрессии Т-клеток путем предотвращения пролиферации за счет вмешательства в ее путь, аналогично трихостатину и вориностату. Другим эффектом VPA является предотвращение пролиферации макрофагов и лимфоцитов в спинном мозге крыс с рассеянным склерозом. В настоящее время ингибиторы HDAC не используются для облегчения симптомов у пациентов с рассеянным склерозом; однако некоторые из них в настоящее время проходят доклинические испытания. [121]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ «Информационный бюллетень о заболеваниях двигательных нейронов: Национальный институт неврологических расстройств и инсульта (NINDS)» . Архивировано из оригинала 13 апреля 2014 г. Проверено 9 августа 2019 г.
- ^ Интернет-менделевское наследование у человека (OMIM): 600882 Болезнь Шарко-Мари-Тута, аксональная, тип 2B; СМТ2Б - 600882
- ^ Сгирланзони А., Парейсон Д., Лаурия Г. (июнь 2005 г.). «Заболевания сенсорных нейронов». обзор. «Ланцет». Неврология . 4 (6): 349–61. дои : 10.1016/S1474-4422(05)70096-X . ПМИД 15907739 . S2CID 35053543 .
- ^ Голл М.Г., Бестор Т.Х. (2005). «Эукариотические цитозинметилтрансферазы». Ежегодный обзор биохимии . 74 : 481–514. doi : 10.1146/annurev.biochem.74.010904.153721 . ПМИД 15952895 .
- ^ Jump up to: а б с д Бернштейн Б.Е., Мейснер А., Ландер Э.С. (февраль 2007 г.). «Эпигеном млекопитающих» . обзор. Клетка . 128 (4): 669–81. дои : 10.1016/j.cell.2007.01.033 . ПМИД 17320505 . S2CID 2722988 .
- ^ Jump up to: а б с Фагихи М.А., Модарреси Ф., Халил А.М., Вуд Д.Э., Сааган Б.Г., Морган Т.Е., Финч С.Э., Сен-Лоран Г., Кенни П.Дж., Валестедт К. (июль 2008 г.). «Экспрессия некодирующей РНК повышается при болезни Альцгеймера и приводит к быстрой прямой регуляции бета-секретазы» . начальный. Природная медицина . 14 (7): 723–30. дои : 10.1038/nm1784 . ПМЦ 2826895 . ПМИД 18587408 .
- ^ Jump up to: а б Урдингио Р.Г., Санчес-Мут СП, Эстеллер М. (ноябрь 2009 г.). «Эпигенетические механизмы неврологических заболеваний: гены, синдромы и методы лечения». «Ланцет». Неврология . 8 (11): 1056–72. дои : 10.1016/S1474-4422(09)70262-5 . ПМИД 19833297 . S2CID 25946604 .
- ^ Пидикайил Дж. (апрель 2013 г.). «Эпигенетические препараты от болезни Альцгеймера» . Британский журнал клинической фармакологии . 75 (4): 1152–3. дои : 10.1111/j.1365-2125.2012.04444.x . ПМЦ 3612735 . ПМИД 22905989 .
- ^ Jump up to: а б Дель Синьор С.Дж., Аманте DJ, Ким Дж., Стэк Э.К., Гудрич С., Кормье К., Смит К., Кудкович М.Е., Ферранте Р.Дж. (апрель 2009 г.). «Комбинированная терапия рилузолом и фенилбутиратом натрия у трансгенных мышей с боковым амиотрофическим склерозом». начальный. Боковой амиотрофический склероз . 10 (2): 85–94. дои : 10.1080/17482960802226148 . ПМИД 18618304 . S2CID 24124109 .
- ^ Jump up to: а б Петри С., Киаи М., Кипиани К., Чен Дж., Калингасан Нью-Йорк, Кроу Дж.П., Бил М.Ф. (апрель 2006 г.). «Аддитивные нейропротекторные эффекты ингибитора деацетилазы гистонов и каталитического антиоксиданта на модели бокового амиотрофического склероза на трансгенных мышах». Нейробиология болезней . 22 (1): 40–9. дои : 10.1016/j.nbd.2005.09.013 . ПМИД 16289867 . S2CID 22794616 .
- ^ Jump up to: а б Кудкович М.Е., Андрес П.Л., Макдональд С.А., Бедлак Р.С., Чоудри Р., Браун Р.Х., Чжан Х., Шенфельд Д.А., Шефнер Дж., Мэтсон С., Мэтсон В.Р., Ферранте Р.Дж. (апрель 2009 г.). «Фаза 2 исследования фенилбутирата натрия при БАС». начальный. Боковой амиотрофический склероз . 10 (2): 99–106. дои : 10.1080/17482960802320487 . ПМИД 18688762 . S2CID 12390136 .
- ^ Jump up to: а б Пайперс С., Велдинк Дж. Х., де Йонг С.В., ван дер Твил И., ван дер Пол В.Л., Уйтендал Э.В., Шелхаас Х.Дж., Шеффер Х., де Виссер М., де Йонг Дж.М., Вокке Дж.Х., Гроеневельд Г.Дж., ван ден Берг Л.Х. (август 2009 г.) ). «Рандомизированное последовательное исследование вальпроевой кислоты при боковом амиотрофическом склерозе». начальный. Анналы неврологии . 66 (2): 227–34. дои : 10.1002/ana.21620 . ПМИД 19743466 . S2CID 44949619 .
- ^ Jump up to: а б с Ю Ю, Ко КП (сентябрь 2011 г.). «Лечение трихостатином А, начатое после начала заболевания, задерживает прогрессирование заболевания и увеличивает выживаемость на мышиной модели бокового амиотрофического склероза». начальный. Экспериментальная неврология . 231 (1): 147–59. doi : 10.1016/j.expneurol.2011.06.003 . ПМИД 21712032 . S2CID 42608157 .
- ^ Jump up to: а б Фишер А., Сананбенези Ф., Ван Х., Доббин М., Цай Л.Х. (май 2007 г.). «Восстановление обучения и памяти связано с ремоделированием хроматина». начальный. Природа . 447 (7141): 178–82. Бибкод : 2007Natur.447..178F . дои : 10.1038/nature05772 . PMID 17468743 . S2CID 36395789 .
- ^ Jump up to: а б с Рикобараса А., Квадрат-Уивер М., Фреймворк С., Перес-Отумн I, Гарсия-Оста А. (май 2012 г.). «Фенилбутират спасает дендритную потерю позвоночника, связанную с дефицитом памяти на мышиной модели болезни Альцгеймера». начальный. Гиппокамп . 22 (5): 1040–50. дои : 10.1002/бегемот.20883 . ПМИД 21069780 . S2CID 20052391 .
- ^ Jump up to: а б Говиндараджан Н., Агис-Бальбоа Р.К., Уолтер Дж., Сананбенеси Ф., Фишер А. (2011). «Бутират натрия улучшает функцию памяти на мышиной модели болезни Альцгеймера при введении на поздней стадии прогрессирования заболевания». начальный. Журнал болезни Альцгеймера . 26 (1): 187–97. дои : 10.3233/JAD-2011-110080 . ПМИД 21593570 .
- ^ Jump up to: а б Килгор М., Миллер К.А., Фасс Д.М., Хенниг К.М., Хаггарти С.Дж., Суэтт Дж.Д., Рамбо Дж. (март 2010 г.). «Ингибиторы деацетилаз гистонов класса 1 устраняют дефицит контекстуальной памяти на мышиной модели болезни Альцгеймера» . начальный. Нейропсихофармакология . 35 (4): 870–80. дои : 10.1038/нпп.2009.197 . ПМК 3055373 . ПМИД 20010553 .
- ^ Jump up to: а б Фрэнсис Й.И., Фа М., Ашраф Х., Чжан Х., Станишевский А., Латчман Д.С., Арансио О (2009). «Нарушение регуляции ацетилирования гистонов в мышиной модели болезни Альцгеймера APP/PS1» . Журнал болезни Альцгеймера . 18 (1): 131–9. дои : 10.3233/JAD-2009-1134 . ПМЦ 8962655 . ПМИД 19625751 .
- ^ Jump up to: а б с д и Стеффан Дж.С., Бодай Л., Паллос Дж., Поулман М., МакКэмпбелл А., Апостол Б.Л., Казанцев А., Шмидт Е., Чжу Ю.З., Гринвальд М., Курокава Р., Хаусман Д.Е., Джексон Г.Р., Марш Дж.Л., Томпсон Л.М. (октябрь 2001 г.). «Ингибиторы гистондеацетилазы останавливают полиглутамин-зависимую нейродегенерацию у дрозофилы» . начальный. Природа . 413 (6857): 739–43. Бибкод : 2001Natur.413..739S . дои : 10.1038/35099568 . ПМИД 11607033 . S2CID 4419980 .
- ^ Jump up to: а б Гардиан Дж., Браун С.Э., Чой Д.К., Кливени П., Грегорио Дж., Кубилус Дж.К., Рю Х., Лэнгли Б., Ратан Р.Р., Ферранте Р.Дж., Бил М.Ф. (январь 2005 г.). «Нейропротекторное действие фенилбутирата на модели болезни Хантингтона на трансгенных мышах N171-82Q» . начальный. Журнал биологической химии . 280 (1): 556–63. дои : 10.1074/jbc.M410210200 . ПМИД 15494404 .
- ^ Jump up to: а б Домпьер Ж.П., Годен Ж.Д., Шаррен Б.К., Кордельер Ф.П., Кинг С.Дж., Умберт С., Сауду Ф. (март 2007 г.). «Ингибирование гистонов деацетилазы 6 компенсирует дефицит транспорта при болезни Хантингтона за счет увеличения ацетилирования тубулина» . начальный. Журнал неврологии . 27 (13): 3571–83. doi : 10.1523/JNEUROSCI.0037-07.2007 . ПМК 6672116 . ПМИД 17392473 .
- ^ Jump up to: а б Чжоу В., Беркури К., Каммиски Дж., Луонг Н., Лебин Дж., Фрид Ч.Р. (апрель 2011 г.). «Фенилбутират повышает регуляцию белка DJ-1 и защищает нейроны в клеточной культуре и на животных моделях болезни Паркинсона» . начальный. Журнал биологической химии . 286 (17): 14941–51. дои : 10.1074/jbc.M110.211029 . ПМК 3083206 . ПМИД 21372141 .
- ^ Jump up to: а б Рэйн П., Шилдс Дж., Хеффернан М., Го Ю., Акбарян С., Кинг Дж. А. (июнь 2012 г.). «Ингибитор деацетилазы гистонов, бутират натрия, облегчает когнитивный дефицит на премоторной стадии БП». начальный. Нейрофармакология . 62 (7): 2409–12. doi : 10.1016/j.neuropharm.2012.01.026 . ПМИД 22353286 . S2CID 23078279 .
- ^ Jump up to: а б Сен-Лоран Р., О'Брайен Л.М., Ахмад С.Т. (август 2013 г.). «Бутират натрия улучшает двигательные нарушения и раннюю смертность в модели болезни Паркинсона, вызванной ротеноном» . начальный. Нейронаука . 246 : 382–90. doi : 10.1016/j.neuroscience.2013.04.037 . ПМК 3721507 . ПМИД 23623990 .
- ^ Jump up to: а б Монти Б., Гатта В., Пиретти Ф., Рафаэлли СС, Виргили М., Контестабиле А. (февраль 2010 г.). «Вальпроевая кислота оказывает нейропротекторное действие на ротеноновой крысиной модели болезни Паркинсона: участие альфа-синуклеина». начальный. Исследования нейротоксичности . 17 (2): 130–41. дои : 10.1007/s12640-009-9090-5 . ПМИД 19626387 . S2CID 40159513 .
- ^ Jump up to: а б с д Контопулос Э., Парвин Дж.Д., Фиани М.Б. (октябрь 2006 г.). «Альфа-синуклеин действует в ядре, ингибируя ацетилирование гистонов и способствуя нейротоксичности» . начальный. Молекулярная генетика человека . 15 (20): 3012–23. дои : 10.1093/hmg/ddl243 . ПМИД 16959795 .
- ^ Jump up to: а б Риссланд М., Брихта Л., Ханен Э., Вирт Б. (август 2006 г.). «Бензамид M344, новый ингибитор гистондеацетилазы, значительно увеличивает уровни РНК/белка SMN2 в клетках спинальной мышечной атрофии». начальный. Генетика человека . 120 (1): 101–10. дои : 10.1007/s00439-006-0186-1 . ПМИД 16724231 . S2CID 24804136 .
- ^ Jump up to: а б с Андреасси С, Анжелоцци С, Тициано Ф.Д., Витали Т, Де Винченци Е, Бонинсенья А, Вилланова М, Бертини Е, Пини А, Нери Г, Браге С (январь 2004 г.). «Фенилбутират увеличивает экспрессию SMN in vitro: актуальность для лечения спинальной мышечной атрофии» . Европейский журнал генетики человека . 12 (1): 59–65. дои : 10.1038/sj.ejhg.5201102 . ПМИД 14560316 .
- ^ Jump up to: а б с Меркури Е, Бертини Е, Мессина С, Солари А, Д'Амико А, Анжелоцци С, Баттини Р, Берардинелли А, Боффи П, Бруно С, Чини С, Колитто Ф, Кинали М, Минетти С, Монджини Т, Моранди Л, Нери Г., Орчези С., Пане М., Пелличчони М., Пини А., Тициано Ф.Д., Вилланова М., Вита Г., Браге С. (январь 2007 г.). «Рандомизированное двойное слепое плацебо-контролируемое исследование фенилбутирата при спинальной мышечной атрофии». начальный. Неврология . 68 (1): 51–5. дои : 10.1212/01.wnl.0000249142.82285.d6 . ПМИД 17082463 . S2CID 30429093 .
- ^ Jump up to: а б Браге К., Витали Т., Тициано Ф.Д., Анжелоцци К., Пинто А.М., Борго Ф., Москато У., Бертини Э., Меркури Э., Нери Дж. (февраль 2005 г.). «Фенилбутират увеличивает экспрессию гена SMN у пациентов со спинальной мышечной атрофией» . начальный. Европейский журнал генетики человека . 13 (2): 256–9. дои : 10.1038/sj.ejhg.5201320 . ПМИД 15523494 .
- ^ Jump up to: а б Самнер С.Дж., Хьюн Т.Н., Марковиц Дж.А., Перхак Дж.С., Хилл Б., Куверт Д.Д., Шусслер К., Чен X, Джареки Дж., Бергес А.Х., Тейлор Дж.П., Фишбек К.Х. (ноябрь 2003 г.). «Вальпроевая кислота повышает уровень SMN в клетках пациентов со спинальной мышечной атрофией». начальный. Анналы неврологии . 54 (5): 647–54. дои : 10.1002/ана.10743 . ПМИД 14595654 . S2CID 7983521 .
- ^ Jump up to: а б Брихта Л., Хофманн Ю., Ханен Э., Зибзенрубль Ф.А., Рашке Х., Блюмке И., Эйюпоглу И.Ю., Вирт Б. (октябрь 2003 г.). «Вальпроевая кислота повышает уровень белка SMN2: хорошо известный препарат как потенциальное средство лечения спинальной мышечной атрофии» . начальный. Молекулярная генетика человека . 12 (19): 2481–9. дои : 10.1093/hmg/ddg256 . ПМИД 12915451 .
- ^ Jump up to: а б Цай Л.К., Цай М.С., Линь ТБ, Хву В.Л., Ли Х (ноябрь 2006 г.). «Создание стандартизированного протокола терапевтического тестирования спинальной мышечной атрофии». начальный. Нейробиология болезней . 24 (2): 286–95. дои : 10.1016/j.nbd.2006.07.004 . ПМИД 16952456 . S2CID 31974628 .
- ^ Jump up to: а б Вейль CC, Коннолли AM, Пестронк А. (август 2006 г.). «Вальпроат может улучшить силу и функциональность у пациентов со спинальной мышечной атрофией III/IV типа». начальный. Неврология . 67 (3): 500–1. дои : 10.1212/01.wnl.0000231139.26253.d0 . ПМИД 16775228 . S2CID 13138072 .
- ^ Jump up to: а б Пиперс С., Коббен Дж.М., Содаар П., Янсен М.Д., Вадман Р.И., Мистер-Делвер А., Poll-The BT, Лемминк Х.Х., Вокке Дж.Х., ван дер Пол В.Л., ван ден Берг Л.Х. (август 2011 г.). «Количественное определение белка SMN в лейкоцитах пациентов со спинальной мышечной атрофией: эффекты лечения вальпроевой кислотой» . начальный. Журнал неврологии, нейрохирургии и психиатрии . 82 (8): 850–2. дои : 10.1136/jnnp.2009.200253 . ПМИД 20551479 . S2CID 27844635 .
- ^ Jump up to: а б Свобода К.Дж., Скотт С.Б., Кроуфорд Т.О., Симард Л.Р., Рейна С.П., Кросселл К.Дж., Асади Г., Эльшейк Б., Шрот М.К., Д'Анжу Г., ЛаСалль Б., Приор Т.В., Соренсон С.Л., Мачульски Дж.А., Бромберг М.Б., Чан Г.М., Кисель Дж.Т. (август 2010 г.). «Исследование SMA CARNI-VAL, часть I: двойное слепое рандомизированное плацебо-контролируемое исследование L-карнитина и вальпроевой кислоты при спинальной мышечной атрофии» . начальный. ПЛОС ОДИН . 5 (8): е12140. Бибкод : 2010PLoSO...512140S . дои : 10.1371/journal.pone.0012140 . ПМЦ 2924376 . ПМИД 20808854 .
- ^ Jump up to: а б Дарбар И.А., Плаггерт П.Г., Ресенде М.Б., Занотели Э., Рид Калифорнийский университет (март 2011 г.). «Оценка мышечной силы и двигательных способностей у детей со спинальной мышечной атрофией II и III типов, получавших вальпроевую кислоту» . начальный. БМК Неврология . 11:36 . дои : 10.1186/1471-2377-11-36 . ПМК 3078847 . ПМИД 21435220 .
- ^ Jump up to: а б Нарвер Х.Л., Конг Л., Бернетт Б.Г., Чо Д.В., Бош-Марсе М., Тайе А.А., Экхаус М.А., Самнер С.Дж. (октябрь 2008 г.). «Устойчивое улучшение состояния мышей со спинальной мышечной атрофией, получавших трихостатин А плюс питание» . начальный. Анналы неврологии . 64 (4): 465–70. дои : 10.1002/ana.21449 . ПМЦ 10103738 . ПМИД 18661558 . S2CID 5595968 .
- ^ Jump up to: а б Авила А.М., Бернетт Б.Г., Тайе А.А., Габанелла Ф., Найт М.А., Хартенштейн П., Чизман З., Ди Просперо Н.А., Пеллиццони Л., Фишбек К.Х., Самнер С.Дж. (март 2007 г.). «Трихостатин А увеличивает экспрессию SMN и выживаемость на мышиной модели спинальной мышечной атрофии» . начальный. Журнал клинических исследований . 117 (3): 659–71. дои : 10.1172/JCI29562 . ПМК 1797603 . ПМИД 17318264 .
- ^ Jump up to: а б Ханен Э, Эйюпоглу И.Ю., Брихта Л., Хаастерт К., Транкле С., Зибценрюбль Ф.А., Риссланд М., Хёлькер И., Клаус П., Ромстёк Дж., Буслей Р., Вирт Б., Блюмке И. (июль 2006 г.). «Оценка ингибиторов гистондеацетилазы второго поколения in vitro и ex vivo для лечения спинальной мышечной атрофии» . начальный. Журнал нейрохимии . 98 (1): 193–202. дои : 10.1111/j.1471-4159.2006.03868.x . ПМИД 16805808 .
- ^ Jump up to: а б с Керночан Л.Е., Руссо М.Л., Вудлинг Н.С., Хьюн Т.Н., Авила А.М., Фишбек К.Х., Самнер С.Дж. (май 2005 г.). «Роль ацетилирования гистонов в экспрессии гена SMN» . начальный. Молекулярная генетика человека . 14 (9): 1171–82. дои : 10.1093/hmg/ddi130 . ПМИД 15772088 .
- ^ Jump up to: а б Риссланд М., Акерманн Б., Фёрстер А., Якубик М., Хауке Дж., Гарбес Л., Фрицше И., Менде Ю., Блюмке И., Ханен Э., Вирт Б. (апрель 2010 г.). «SAHA улучшает фенотип СМА в двух моделях спинальной мышечной атрофии на мышах» . начальный. Молекулярная генетика человека . 19 (8): 1492–506. дои : 10.1093/hmg/ddq023 . ПМИД 20097677 .
- ^ Дьюи CM, Ченик Б, Сефтон CF, Джонсон BA, Герц Дж, Ю Г (июнь 2012 г.). «Агрегация TDP-43 при нейродегенерации: являются ли стрессовые гранулы ключом?» . обзор. Исследования мозга . 1462 : 16–25. дои : 10.1016/j.brainres.2012.02.032 . ПМЦ 3372581 . ПМИД 22405725 .
- ^ Полимениду М., Лажье-Туренн К., Хатт КР, Беннетт К.Ф., Кливленд Д.В., Йео Г.В. (июнь 2012 г.). «Неправильная регуляция процессинга РНК при боковом амиотрофическом склерозе» . обзор. Исследования мозга . 1462 : 3–15. дои : 10.1016/j.brainres.2012.02.059 . ПМК 3707312 . ПМИД 22444279 .
- ^ Руо С., Йокич Н., Мбеби С., Бутилье С., Леффлер Дж.П., Бутилье А.Л. (декабрь 2003 г.). «Критическая потеря активности ацетилазы гистонов CBP/p300 каспазой-6 во время нейродегенерации» . начальный. Журнал ЭМБО . 22 (24): 6537–49. дои : 10.1093/emboj/cdg615 . ПМК 291810 . ПМИД 14657026 .
- ^ Баттистини С., Риччи С., Лотти Э.М., Бениньи М., Гальярди С., Зукко Р., Бондавалли М., Марчелло Н., Черони М., Середа С. (июнь 2010 г.). «Тяжелый семейный БАС с новой мутацией экзона 4 (L106F) в гене SOD1». начальный. Журнал неврологических наук . 293 (1–2): 112–5. дои : 10.1016/j.jns.2010.03.009 . ПМИД 20385392 . S2CID 24895265 .
- ^ Брюйн Л.И., Houseweart MK, Като С., Андерсон К.Л., Андерсон С.Д., Охама Э., Реоме А.Г., Скотт Р.В., Кливленд Д.В. (сентябрь 1998 г.). «Агрегация и токсичность двигательных нейронов мутанта SOD1, связанного с ALS, независимого от SOD1 дикого типа». начальный. Наука . 281 (5384): 1851–4. Бибкод : 1998Sci...281.1851B . дои : 10.1126/science.281.5384.1851 . ПМИД 9743498 .
- ^ Фурукава Ю., Фу Р., Дэн Х.С., Сиддик Т., О'Халлоран ТВ (май 2006 г.). «Дисульфидно-сшитый белок представляет собой значительную долю агрегатов Cu, Zn-супероксиддисмутазы, связанных с БАС, в спинном мозге модельных мышей» . начальный. Труды Национальной академии наук Соединенных Штатов Америки . 103 (18): 7148–53. Бибкод : 2006PNAS..103.7148F . дои : 10.1073/pnas.0602048103 . ПМЦ 1447524 . ПМИД 16636274 .
- ^ Бойле С., Ванде Вельде С., Кливленд Д.В. (октябрь 2006 г.). «АЛС: заболевание двигательных нейронов и их ненейрональных соседей» . обзор. Нейрон . 52 (1): 39–59. дои : 10.1016/j.neuron.2006.09.018 . ПМИД 17015226 . S2CID 12968143 .
- ^ Кудкович М.Е., Маккенна-Ясек Д., Сапп П.Е., Чин В., Геллер Б., Хайден Д.Л., Шенфельд Д.А., Хослер Б.А., Хорвиц Х.Р., Браун Р.Х. (февраль 1997 г.). «Эпидемиология мутаций супероксиддисмутазы при боковом амиотрофическом склерозе» . начальный. Анналы неврологии . 41 (2): 210–21. дои : 10.1002/ana.410410212 . ПМИД 9029070 . S2CID 25595595 .
- ^ Jump up to: а б Тодд Т.В., Петручелли Л. (август 2016 г.). «Понимание патогенетических механизмов расширения повторов открытой рамки считывания 72 (C9orf72) хромосомы 9» . обзор. Журнал нейрохимии . 138 (Приложение 1): 145–62. дои : 10.1111/jnc.13623 . ПМИД 27016280 .
- ^ Йошимура С., Герондопулос А., Линфорд А., Ригден DJ, Барр Ф.А. (октябрь 2010 г.). «Общесемейная характеристика коэффициентов обмена Rab GDP-GTP в домене DENN» . начальный. Журнал клеточной биологии . 191 (2): 367–81. дои : 10.1083/jcb.201008051 . ПМЦ 2958468 . ПМИД 20937701 .
- ^ Дэн Х.Х., Чен В., Хонг С.Т., Бойкот К.М., Горри Г.Х., Сиддик Н. и др. (август 2011 г.). «Мутации в UBQLN2 вызывают доминантный Х-сцепленный БАС у юношей и взрослых, а также БАС/деменцию» . начальный. Природа . 477 (7363): 211–5. Бибкод : 2011Natur.477..211D . дои : 10.1038/nature10353 . ПМК 3169705 . ПМИД 21857683 .
- ^ Руо С., Леффлер Дж.П., Бутилье А.Л. (сентябрь 2004 г.). «Нацеливание на потерю функции CREB-связывающего белка (CBP) как терапевтическая стратегия при неврологических расстройствах». обзор. Биохимическая фармакология . 68 (6): 1157–64. дои : 10.1016/j.bcp.2004.05.035 . ПМИД 15313413 .
- ^ Jump up to: а б Рю Х., Смит К., Камело С.И., Каррерас И., Ли Дж., Иглесиас А.Х., Дангонд Ф., Кормье К.А., Кудкович М.Е., Браун Р.Х., Ферранте Р.Дж. (июнь 2005 г.). «Фенилбутират натрия продлевает выживаемость и регулирует экспрессию антиапоптотических генов у трансгенных мышей с боковым амиотрофическим склерозом» . начальный. Журнал нейрохимии . 93 (5): 1087–98. дои : 10.1111/j.1471-4159.2005.03077.x . ПМИД 15934930 .
- ^ Коркоран Л.Дж., Митчисон Т.Дж., Лю Кью (март 2004 г.). «Новое действие ингибиторов гистондеацетилазы на модели белково-агресомного заболевания» . начальный. Современная биология . 14 (6): 488–92. Бибкод : 2004CBio...14..488C . дои : 10.1016/j.cub.2004.03.003 . ПМИД 15043813 . S2CID 6465499 .
- ^ Крочемор С, Виргили М, Бонамасса Б, Канистро Д, Пена-Альтамира Е, Паолини М, Контестабиле А (апрель 2009 г.). «Длительное диетическое введение вальпроевой кислоты не влияет, в то время как ретиноевая кислота уменьшает продолжительность жизни мышей G93A, модели бокового амиотрофического склероза». начальный. Мышцы и нервы . 39 (4): 548–52. дои : 10.1002/mus.21260 . ПМИД 19296491 . S2CID 26101773 .
- ^ Руо С., Пантелеева И., Рене Ф., Гонсалес де Агилар Х.Л., Эчанис-Лагуна А., Дюпюи Л., Менгер Ю., Бутилье А.Л., Леффлер Дж.П. (май 2007 г.). «Вальпроат натрия оказывает нейропротекторное действие in vivo посредством механизмов, зависящих от белка-связывания CREB, но не улучшает выживаемость на мышиной модели бокового амиотрофического склероза» . начальный. Журнал неврологии . 27 (21): 5535–45. doi : 10.1523/JNEUROSCI.1139-07.2007 . ПМК 6672753 . ПМИД 17522299 .
- ^ Бжустович Л.М., Ленер Т., Кастилья Л.Х., Пенчасзаде Г.К., Вильгельмсен К.К., Дэниелс Р., Дэвис К.Э., Лепперт М., Цитер Ф., Вуд Д. (апрель 1990 г.). «Генетическое картирование хронической спинальной мышечной атрофии, возникшей в детстве, хромосомы 5q11.2–13.3». начальный. Природа . 344 (6266): 540–1. Бибкод : 1990Natur.344..540B . дои : 10.1038/344540a0 . ПМИД 2320125 . S2CID 4259327 .
- ^ Прайор Т.В., Крайнер А.Р., Хуа Ю., Свобода К.Дж., Снайдер ПК, Бриджмен С.Дж., Бергес А.Х., Кисел Дж.Т. (сентябрь 2009 г.). «Положительный модификатор спинальной мышечной атрофии в гене SMN2» . начальный. Американский журнал генетики человека . 85 (3): 408–13. дои : 10.1016/j.ajhg.2009.08.002 . ПМЦ 2771537 . ПМИД 19716110 .
- ^ Шремл Дж., Риссланд М., Патерно М., Гарбес Л., Россбах К., Акерманн Б., Кремер Дж., Сомерс Э., Парсон Ш., Хеллер Р., Беркессель А., Штернер-Кок А., Вирт Б. (июнь 2013 г.). «У мышей с тяжелой формой СМА наблюдаются нарушения работы органов, которые не могут быть устранены терапией HDACi JNJ-26481585» . начальный. Европейский журнал генетики человека . 21 (6): 643–52. дои : 10.1038/ejhg.2012.222 . ПМЦ 3658191 . ПМИД 23073311 .
- ^ Лазаридис К., Цартос С.Дж. (2020). «Специфичность аутоантител при миастении гравис: значение для улучшения диагностики и терапии» . Границы в иммунологии . 11 : 212. дои : 10.3389/fimmu.2020.00212 . ISSN 1664-3224 . ПМК 7033452 . ПМИД 32117321 .
- ^ Jump up to: а б Авидан Н., Ле Панс Р., Берри-Акнин С., Миллер А. (01.08.2014). «Генетическая основа миастении. Всесторонний обзор» . Журнал аутоиммунитета . Миастения Гравис: всесторонний обзор. 52 : 146–153. дои : 10.1016/j.jaut.2013.12.001 . ISSN 0896-8411 . ПМИД 24361103 .
- ^ «Миастения Гравис» . www.hopkinsmedicine.org . 08.08.2021 . Проверено 18 мая 2022 г.
- ^ Гольфинопулу Р., Папагеоргиу Л., Эфтимиаду А., Бакопулу Ф., Хрусос Г.П., Элиопулос Э., Влачакис Д. (01.07.2021). «Клинические геномные, фенотипические и эпигенетические данные о патологии, аутоиммунитете и контроле веса пациентов с миастенией гравис (обзор)» . Отчеты о молекулярной медицине . 24 (1): 1–9. дои : 10.3892/ммр.2021.12151 . ISSN 1791-2997 . ПМИД 34225443 . S2CID 235744938 .
- ^ Jump up to: а б с Ван Л, Чжан Л (2020). «Новая роль дисрегуляции микроРНК при миастении гравис» . Границы в неврологии . 14 : 507. дои : 10.3389/fnins.2020.00507 . ISSN 1662-453X . ПМЦ 7253668 . ПМИД 32508584 .
- ^ Jump up to: а б Шрёбель П., Розенвальд А., Байерсдорф Н., Керкау Т., Элерт О., Мурумяги А., Силланпяя Н., Петерсон П., Хуммель В., Рикманн П., Бурек С. (2004). «Селективная потеря регуляторных Т-клеток в тимомах» . Анналы неврологии . 56 (6): 901–904. дои : 10.1002/ана.20340 . ISSN 0364-5134 . ПМИД 15562414 . S2CID 43398892 .
- ^ Крон М.А., Майяр С., Делиль Ф., Самсон Н., Трюффо Ф., Фоти М., Фадель Э., Гихайр Дж., Берри-Акнин С., Ле Панс Р. (2018). «Анализ экспрессии микроРНК в тимусе пациентов с миастенией гравис открывает новые возможности для исследований» . Обзоры аутоиммунитета . 17 (6): 588–600. дои : 10.1016/j.autrev.2018.01.008 . ISSN 1873-0183 . ПМИД 29655674 . S2CID 4882702 .
- ^ Лю XF, Ван RQ, Ху Б, Ло MC, Цзэн QM, Чжоу Х, Хуан К, Донг XH, Луо YB, Ло Чж, Ян Х (01 марта 2016 г.). «МиР-15а способствует аномальному иммунному ответу при миастении, нацеливаясь на CXCL10» . Клиническая иммунология . 164 : 106–113. дои : 10.1016/j.clim.2015.12.009 . ISSN 1521-6616 . ПМИД 26845678 .
- ^ Лу Дж, Ян М, Ван Ю, Чжан Дж, Ян Х, Тянь Фф, Чжоу В, Чжан Н, Ли Дж (25 октября 2013 г.). «Измененная экспрессия миР-146a при миастении» . Письма по неврологии . 555 : 85–90. дои : 10.1016/j.neulet.2013.09.014 . ISSN 0304-3940 . ПМИД 24036458 . S2CID 5317928 .
- ^ Ванюшин Б.Ф. (2004). «Ферментативное метилирование ДНК является эпигенетическим контролем генетических функций клетки» . Биохимия (Москва) . 70 (5): 488–499. дои : 10.1007/s10541-005-0143-y . ПМИД 15948703 . S2CID 25973184 .
- ^ Фанг Т.К., Ян СиДжей, Ду Дж (2018). «Метилирование CTLA-4 регулирует патогенез миастении и экспрессию родственных цитокинов» . Лекарство . 97 (18): e0620. дои : 10.1097/MD.0000000000010620 . ISSN 0025-7974 . ПМК 6393147 . ПМИД 29718870 .
- ^ Крон М.А., Гильошон Э, Куснер Л., Ле Панс Р. (10 июня 2020 г.). «Роль микроРНК в нормальном состоянии и миастении гравис тимуса» . Границы в иммунологии . 11 : 1074. дои : 10.3389/fimmu.2020.01074 . ISSN 1664-3224 . ПМЦ 7297979 . ПМИД 32587589 .
- ^ Фанг Т.К., Ян СиДжей, Ду Дж (2018). «Метилирование CTLA-4 регулирует патогенез миастении и экспрессию родственных цитокинов» . Лекарство . 97 (18): e0620. дои : 10.1097/MD.0000000000010620 . ISSN 1536-5964 . ПМК 6393147 . ПМИД 29718870 .
- ^ Jump up to: а б Сюй С, Ван Т, Лу Икс, Чжан Х, Лю Л, Конг Икс, Ли С, Ван Икс, Гао Х, Ван Дж, Ван Л (2021). «Идентификация LINC00173 при миастении гравис путем интеграционного анализа аберрантно метилированных и дифференциально экспрессируемых генов и сетей ceRNA» . Границы генетики . 12 : 726751. doi : 10.3389/fgene.2021.726751 . ISSN 1664-8021 . ПМЦ 8481885 . ПМИД 34603387 .
- ^ «Ген HLA-DRB1: MedlinePlus Genetics» . medlineplus.gov . Проверено 12 апреля 2022 г.
- ^ Де Ваал Малефит Р., Абрамс Дж., Беннетт Б., Фигдор К.Г., Де Врис Дж.Э. (01.11.1991). «Интерлейкин 10 (IL-10) ингибирует синтез цитокинов моноцитами человека: ауторегуляторная роль IL-10, продуцируемого моноцитами» . Журнал экспериментальной медицины . 174 (5): 1209–1220. дои : 10.1084/jem.174.5.1209 . ISSN 0022-1007 . ПМК 2119001 . ПМИД 1940799 .
- ^ Фуджи Си, Симидзу К., Симидзу Т., Лотце М.Т. (01.10.2001). «Интерлейкин-10 способствует поддержанию эффекторной функции противоопухолевых CD8+ Т-клеток in situ» . Кровь . 98 (7): 2143–2151. дои : 10.1182/blood.V98.7.2143 . ISSN 0006-4971 . ПМИД 11568001 .
- ^ Пунга А.Р., Андерссон М., Алимохаммади М., Пунга Т. (15 сентября 2015 г.). «Специфические для заболевания признаки циркулирующих миР-150-5p и миР-21-5p у пациентов с миастенией» . Журнал неврологических наук . 356 (1): 90–96. дои : 10.1016/j.jns.2015.06.019 . ISSN 0022-510X . ПМИД 26095457 . S2CID 9099858 .
- ^ Беннетт Д.А., Ю Л., Ян Дж., Шривастава Г.П., Обин С., Де Ягер П.Л. (январь 2015 г.). «Эпигеномика болезни Альцгеймера» . обзор. Трансляционные исследования . 165 (1): 200–20. дои : 10.1016/j.trsl.2014.05.006 . ПМЦ 4233194 . ПМИД 24905038 .
- ^ Мастроени Д., Макки А., Гровер А., Роджерс Дж., Коулман П.Д. (август 2009 г.). «Эпигенетические различия корковых нейронов от пары монозиготных близнецов, дискордантных по болезни Альцгеймера» . начальный. ПЛОС ОДИН . 4 (8): е6617. Бибкод : 2009PLoSO...4.6617M . дои : 10.1371/journal.pone.0006617 . ПМК 2719870 . ПМИД 19672297 .
- ^ Шулиарас Л., Мастроени Д., Дельво Э., Гровер А., Кенис Г., Хоф П.Р., Стейнбуш Х.В., Коулман П.Д., Руттен Б.П., ван ден Хове Д.Л. (сентябрь 2013 г.). «Последовательное снижение глобального метилирования и гидроксиметилирования ДНК в гиппокампе пациентов с болезнью Альцгеймера» . начальный. Нейробиология старения . 34 (9): 2091–9. doi : 10.1016/j.neurobiolaging.2013.02.021 . ПМЦ 3955118 . ПМИД 23582657 .
- ^ Мастроени Д., Гровер А., Дельво Э., Уайтсайд С., Коулман П.Д., Роджерс Дж. (декабрь 2010 г.). «Эпигенетические изменения при болезни Альцгеймера: снижение метилирования ДНК» . начальный. Нейробиология старения . 31 (12): 2025–37. doi : 10.1016/j.neurobiolaging.2008.12.005 . ПМЦ 2962691 . ПМИД 19117641 .
- ^ Бакульский К.М., Долиной Д.С., Сартор М.А., Полсон Х.Л., Конен Дж.Р., Либерман А.П., Альбин Р.Л., Ху Х., Розек Л.С. (2012). «Различия в метилировании ДНК по всему геному между болезнью Альцгеймера с поздним началом и когнитивно нормальным контролем в лобной коре человека» . Журнал болезни Альцгеймера . 29 (3): 571–88. дои : 10.3233/JAD-2012-111223 . ПМЦ 3652332 . ПМИД 22451312 .
- ^ Jump up to: а б с д Рао Дж.С., Келешян В.Л., Кляйн С., Рапопорт С.И. (июль 2012 г.). «Эпигенетические модификации лобной коры головного мозга у пациентов с болезнью Альцгеймера и биполярным расстройством» . начальный. Трансляционная психиатрия . 2 (7): е132. дои : 10.1038/tp.2012.55 . ПМЦ 3410632 . ПМИД 22760556 .
- ^ Ван Ю, Чжан JX, Ду XX, Чжао Л, Тянь Ц, Чжу LQ, Ван Ш, Ван JZ (сентябрь 2008 г.). «Временная корреляция дефицита памяти с поражениями, подобными болезни Альцгеймера, вызванными активацией киназы гликогенсинтазы-3» . Журнал нейрохимии . 106 (6): 2364–74. дои : 10.1111/j.1471-4159.2008.05578.x . ПМИД 18643871 .
- ^ Николия В., Фусо А., Кавалларо Р.А., Ди Луцио А., Скарпа С. (2010). «Дефицит витамина B способствует фосфорилированию тау посредством регуляции GSK3beta и PP2A». начальный. Журнал болезни Альцгеймера . 19 (3): 895–907. дои : 10.3233/JAD-2010-1284 . ПМИД 20157245 .
- ^ Бюн Си Джей, Со Джей, Джо С.А., Пак Ю.Дж., Клуг М., Рели М., Пак М.Х., Джо И (январь 2012 г.). «Метилирование ДНК 5'-нетранслируемой области по адресам +298 и +351 подавляет экспрессию BACE1 в микроглиальных клетках BV-2 мыши». начальный. Связь с биохимическими и биофизическими исследованиями . 417 (1): 387–92. дои : 10.1016/j.bbrc.2011.11.123 . ПМИД 22166205 .
- ^ Чен К.Л., Ван С.С., Ян Ю.Ю., Юань Р.Ю., Чен Р.М., Ху С.Дж. (январь 2009 г.). «Эпигенетические эффекты бета-амилоида (1–40) на глобальную ДНК и гены неприлизина в эндотелиальных клетках головного мозга мышей» . начальный. Связь с биохимическими и биофизическими исследованиями . 378 (1): 57–61. дои : 10.1016/j.bbrc.2008.10.173 . ПМИД 19007750 .
- ^ Тоги Х, Абэ Т, Ямазаки К, Мурата Т, Ишизаки Э, Исобе С (июль 1999 г.). «Изменения концентрации 3-нитротирозина в спинномозговой жидкости при старении и у пациентов с болезнью Альцгеймера». начальный. Письма по неврологии . 269 (1): 52–4. дои : 10.1016/S0304-3940(99)00406-1 . ПМИД 10821643 . S2CID 20536297 .
- ^ Чжан К., Шраг М., Крофтон А., Триведи Р., Винтерс Х., Кирш В. (апрель 2012 г.). «Целевая протеомика для количественной оценки ацетилирования гистонов при болезни Альцгеймера» . начальный. Протеомика . 12 (8): 1261–8. дои : 10.1002/pmic.201200010 . ПМК 6812507 . ПМИД 22577027 .
- ^ Грефф Дж., Рей Д., Гуан Дж.С., Ван В.И., Со Дж., Хенниг К.М., Ниланд Т.Дж., Фасс Д.М., Као П.Ф., Кан М., Су СК, Самей А., Джозеф Н., Хаггарти С.Дж., Делалле И., Цай Л.Х. (февраль 2012 г.) ). «Эпигенетическая блокада когнитивных функций в нейродегенеративном мозге» . начальный. Природа . 483 (7388): 222–6. Бибкод : 2012Natur.483..222G . дои : 10.1038/nature10849 . ПМЦ 3498952 . ПМИД 22388814 .
- ^ Пелег С, Сананбенези Ф, Зовойлис А, Буркхардт С, Бахари-Яван С, Агис-Бальбоа Р.К., Кота П, Виттнам Дж.Л., Гоголь-Деринг А, Опиц Л., Салинас-Ристер Г., Деттенхофер М., Канг Х, Фаринелли Л, Чен В., Фишер А. (май 2010 г.). «Измененное ацетилирование гистонов связано с возрастными нарушениями памяти у мышей». начальный. Наука . 328 (5979): 753–6. Бибкод : 2010Sci...328..753P . дои : 10.1126/science.1186088 . ПМИД 20448184 . S2CID 7370920 .
- ^ Фусо А (март 2013 г.). «Золотой век» метилирования ДНК при нейродегенеративных заболеваниях . обзор. Клиническая химия и лабораторная медицина . 51 (3): 523–34. дои : 10.1515/cclm-2012-0618 . ПМИД 23183753 . S2CID 36486849 .
- ^ Хан А.А., Мао С.О., Банвейт С., Джин К., Гринберг Д.А. (ноябрь 2007 г.). «Нейроглобин ослабляет нейротоксичность бета-амилоида in vitro и трансгенный фенотип болезни Альцгеймера in vivo» . начальный. Труды Национальной академии наук Соединенных Штатов Америки . 104 (48): 19114–9. Бибкод : 2007PNAS..10419114K . дои : 10.1073/pnas.0706167104 . ПМК 2141917 . ПМИД 18025470 .
- ^ Чжан В., Тянь З., Ша С., Ченг Л.И., Филипсен С., Тан-Ун К.С. (2011). «Функциональный и последовательностной анализ промоторной области гена нейроглобина человека». начальный. Biochimica et Biophysica Acta (BBA) - Механизмы регуляции генов . 1809 (4–6): 236–44. дои : 10.1016/j.bbagrm.2011.02.003 . ПМИД 21362510 .
- ^ Гуан Дж.С., Хаггарти С.Дж., Джакометти Э., Данненберг Дж.Х., Джозеф Н., Гао Дж., Ниланд Т.Дж., Чжоу Ю., Ван Х, Мазичек Р., Брэднер Дж.Э., ДеПиньо Р.А., Джениш Р., Цай Л.Х. (май 2009 г.). «HDAC2 отрицательно регулирует формирование памяти и синаптическую пластичность» . начальный. Природа . 459 (7243): 55–60. Бибкод : 2009Natur.459...55G . дои : 10.1038/nature07925 . ПМЦ 3498958 . ПМИД 19424149 .
- ^ Jump up to: а б Онлайн-менделевское наследование у человека (OMIM): болезнь Хантингтона - 143100
- ^ Насир Дж., Флореско С.Б., О'Куски Дж.Р., Диверт В.М., Ричман Дж.М., Зейслер Дж., Боровски А., Март Дж.Д., Филлипс А.Г., Хайден М.Р. (июнь 1995 г.). «Направленное разрушение гена болезни Хантингтона приводит к эмбриональной смертности, а также поведенческим и морфологическим изменениям у гетерозигот» . начальный. Клетка . 81 (5): 811–23. дои : 10.1016/0092-8674(95)90542-1 . ПМИД 7774020 . S2CID 16835259 .
- ^ Чен С., Ферроне Ф.А., Ветцель Р. (сентябрь 2002 г.). «Возраст начала болезни Хантингтона связан с зарождением агрегации полиглутамина» . начальный. Труды Национальной академии наук Соединенных Штатов Америки . 99 (18): 11884–9. Бибкод : 2002PNAS...9911884C . дои : 10.1073/pnas.182276099 . ПМК 129363 . ПМИД 12186976 .
- ^ Jump up to: а б с д Рю Х., Ли Дж., Хагерти С.В., Со Б.И., Макалпин С.Е., Кормье К.А., Смит К.М., Ферранте Р.Дж. (декабрь 2006 г.). «Экспрессия гена ESET/SETDB1 и триметилирование гистона H3 (K9) при болезни Хантингтона» . начальный. Труды Национальной академии наук Соединенных Штатов Америки . 103 (50): 19176–81. Бибкод : 2006PNAS..10319176R . дои : 10.1073/pnas.0606373103 . ПМК 1748195 . ПМИД 17142323 .
- ^ Jump up to: а б с Хазеки Н., Цукамото Т., Ядзава И., Кояма М., Хаттори С., Сомеки И., Ивацубо Т., Накамура К., Гото Дж., Канадзава И. (июнь 2002 г.). «Ультраструктура ядерных агрегатов, образованных путем экспрессии расширенного полиглутамина». начальный. Связь с биохимическими и биофизическими исследованиями . 294 (2): 429–40. дои : 10.1016/S0006-291X(02)00498-9 . ПМИД 12051730 .
- ^ Jump up to: а б с Садри-Вакили Г., Бузу Б., Бенн К.Л., Ким М.О., Чавла П., Оверленд Р.П., Глайч К.Е., Ся Э., Цю З., Херш С.М., Кларк Т.В., Йорлинг Г.Дж., Ча Дж.Х. (июнь 2007 г.). «Гистоны, связанные с генами с пониженной регуляцией, гипоацетилированы в моделях болезни Хантингтона» . начальный. Молекулярная генетика человека . 16 (11): 1293–306. дои : 10.1093/hmg/ddm078 . ПМИД 17409194 .
- ^ Хогарт П., Ловречич Л., Крайнц Д. (октябрь 2007 г.). «Фенилбутират натрия при болезни Хантингтона: исследование по подбору дозы». начальный. Двигательные расстройства . 22 (13): 1962–4. дои : 10.1002/mds.21632 . ПМИД 17702032 . S2CID 10846780 .
- ^ Энтрез Ген. «БДНФ» . Национальный центр биотехнологической информации США .
- ^ Ким Дж., Иноуэ К., Исии Дж., Ванти В.Б., Воронов С.В., Мерчисон Э., Хэннон Г., Абелиович А. (август 2007 г.). «Цепь обратной связи микроРНК в дофаминовых нейронах среднего мозга» . начальный. Наука . 317 (5842): 1220–4. Бибкод : 2007Sci...317.1220K . дои : 10.1126/science.1140481 . ПМЦ 2782470 . ПМИД 17761882 .
- ^ Янкович Дж., Чен С., Ле В.Д. (2005). «Роль Nurr1 в развитии дофаминергических нейронов и болезни Паркинсона». обзор. Прогресс нейробиологии . 77 (1–2): 128–38. doi : 10.1016/j.pneurobio.2005.09.001 . ПМИД 16243425 . S2CID 22764367 .
- ^ Доксакис Э (апрель 2010 г.). «Посттранскрипционная регуляция экспрессии альфа-синуклеина с помощью mir-7 и mir-153» . начальный. Журнал биологической химии . 285 (17): 12726–34. дои : 10.1074/jbc.M109.086827 . ПМЦ 2857101 . ПМИД 20106983 .
- ^ Пипер ХК, Эверт Б.О., Каут О., Ридерер П.Ф., Ваха А., Вюлльнер Ю. (декабрь 2008 г.). «Разное метилирование промотора TNF-альфа в коре головного мозга и черной субстанции: последствия для избирательной уязвимости нейронов». начальный. Нейробиология болезней . 32 (3): 521–7. дои : 10.1016/j.nbd.2008.09.010 . ПМИД 18930140 . S2CID 8673158 .
- ^ Моги М., Харада М., Нарабаяши Х., Инагаки Х., Минами М., Нагацу Т. (июнь 1996 г.). «Уровни интерлейкина (IL)-1 бета, IL-2, IL-4, IL-6 и трансформирующего фактора роста-альфа повышены в желудочковой спинномозговой жидкости при ювенильном паркинсонизме и болезни Паркинсона». начальный. Письма по неврологии . 211 (1): 13–6. дои : 10.1016/0304-3940(96)12706-3 . ПМИД 8809836 . S2CID 54279479 .
- ^ Бонш Д., Ленц Б., Корнхубер Дж., Блайх С. (февраль 2005 г.). «Гиперметилирование ДНК промотора альфа-синуклеина у больных алкоголизмом». начальный. НейроОтчет . 16 (2): 167–70. дои : 10.1097/00001756-200502080-00020 . ПМИД 15671870 . S2CID 43289612 .
- ^ Йоваед А., Шмитт И., Каут О., Вюлльнер У. (май 2010 г.). «Метилирование регулирует экспрессию альфа-синуклеина и снижается в мозгу пациентов с болезнью Паркинсона» . начальный. Журнал неврологии . 30 (18): 6355–9. doi : 10.1523/JNEUROSCI.6119-09.2010 . ПМК 6632710 . ПМИД 20445061 .
- ^ Десплатс П., Спенсер Б., Коффи Е., Патель П., Майкл С., Патрик С., Адаме А., Рокенштейн Е., Маслия Е. (март 2011 г.). «Альфа-синуклеин изолирует Dnmt1 из ядра: новый механизм эпигенетических изменений при заболеваниях с тельцами Леви» . начальный. Журнал биологической химии . 286 (11): 9031–7. дои : 10.1074/jbc.C110.212589 . ПМК 3059002 . ПМИД 21296890 .
- ^ Jump up to: а б Оутейро Т.Ф., Контопулос Э., Альтманн С.М., Куфарева И., Стратхерн К.Е., Аморе А.М., Волк С.Б., Максвелл М.М., Роше Дж.К., Маклин П.Дж., Янг А.Б., Абагян Р., Фиани М.Б., Хайман Б.Т., Казанцев А.Г. (июль 2007 г.). «Ингибиторы сиртуина 2 устраняют токсичность, опосредованную альфа-синуклеином, на моделях болезни Паркинсона». начальный. Наука . 317 (5837): 516–9. Бибкод : 2007Sci...317..516O . дои : 10.1126/science.1143780 . PMID 17588900 . S2CID 84493360 .
- ^ Сонг С, Кантасами А., Джин Х., Анантарам В., Кантасами А.Г. (октябрь 2011 г.). «Паракват вызывает эпигенетические изменения, способствуя ацетилированию гистонов в моделях клеточной культуры дофаминергической дегенерации» . начальный. Нейротоксикология . 32 (5): 586–95. дои : 10.1016/j.neuro.2011.05.018 . ПМК 3407036 . ПМИД 21777615 .
- ^ Харрисон И.Ф., Декстер Д.Т. (октябрь 2013 г.). «Эпигенетическое воздействие на деацетилазу гистонов: терапевтический потенциал при болезни Паркинсона?». обзор. Фармакология и терапия . 140 (1): 34–52. doi : 10.1016/j.pharmthera.2013.05.010 . ПМИД 23711791 .
- ^ "Рассеянный склероз" . medlineplus.gov . Проверено 28 марта 2022 г.
- ^ «Рассеянный склероз – симптомы и причины» . Клиника Мэйо . Проверено 28 марта 2022 г.
- ^ Jump up to: а б Грейвс MC, Бентон М., Леа Р.А., Бойл М., Таджури Л., Макартни-Коксон Д., Скотт Р.Дж., Лехнер-Скотт Дж. (12 декабря 2013 г.). «Различия в метилировании локуса HLA-DRB1 в CD4+ Т-клетках связаны с рассеянным склерозом» . Журнал рассеянного склероза . 20 (8): 1033–1041. дои : 10.1177/1352458513516529 . ISSN 1352-4585 . ПМИД 24336351 . S2CID 45718656 .
- ^ Кулар Л., Лю Ю., Рурманн С., Железнякова Г., Марабита Ф., Гомес-Кабреро Д., Джеймс Т., Юинг Э., Линден М., Гурникевич Б., Айнехбанд С. (19 июня 2018 г.). «Метилирование ДНК как медиатор HLA-DRB1*15:01 и защитный вариант при рассеянном склерозе» . Природные коммуникации . 9 (1): 2397. Бибкод : 2018NatCo...9.2397K . дои : 10.1038/s41467-018-04732-5 . ISSN 2041-1723 . ПМК 6008330 . ПМИД 29921915 .
- ^ Jump up to: а б с Кючюкали С.И., Куртунчу М., Чобан А., Чеби М., Тюзюн Э. (2015). «Эпигенетика рассеянного склероза: обновленный обзор» . Нейромолекулярная медицина . 17 (2): 83–96. дои : 10.1007/s12017-014-8298-6 . ISSN 1559-1174 . ПМИД 24652042 . S2CID 9147572 .
- ^ Пидикайил Дж (2016). «Эпигенетические препараты при рассеянном склерозе» . Современная нейрофармакология . 14 (1): 3–9. дои : 10.2174/1570159X13666150211001600 . ISSN 1570-159X . ПМЦ 4787283 . ПМИД 26813117 .