Открытие и разработка ингибиторов АПФ
Открытие перорально неактивного пептида из змеиного яда установило важную роль ангиотензинпревращающего фермента (АПФ) ингибиторов в регулировании артериального давления . Это привело к разработке каптоприла . , первого АПФ ингибитора Когда побочные эффекты каптоприла стали очевидны, были разработаны новые производные. Затем, после открытия двух активных центров АПФ: N-домена и С-домена, началась разработка домен-специфических ингибиторов АПФ. [ 1 ] [ 2 ]
Разработка ингибиторов АПФ первого поколения
[ редактировать ]Разработка нонапептида тепротида ( Glu - Trp - Pro - Arg -Pro- Gln - Ile -Pro-Pro), который первоначально был выделен из яда бразильской гадюки Bothrops jararaca , значительно прояснила важность АПФ при гипертонии . Однако отсутствие пероральной активности ограничивало его терапевтическую полезность. [ 3 ] [ 4 ]
L- бензилянтарная кислота (2(R)-бензил-3-карбоксипионовая кислота) была описана как наиболее мощный ингибитор карбоксипептидазы А в начале 1980-х годов. Авторы группу назвали его аналогом продукта побочного и предположили, что он связывается с активным центром карбоксипептидазы А через сукцинилкарбоксильную и карбонильную группу . Их результаты установили, что L-бензилянтарная кислота связана в одном локусе активного сайта карбоксипептидазы А. Авторы обсудили, но отвергли предположение о том, что карбоксилатная функция может связываться с каталитически функциональным цинка ионом , присутствующим в активном сайте. Однако позже выяснилось, что это так. [ 3 ] [ 5 ] [ 6 ]
Разработка лекарственного средства каптоприл (сульфгидрилы)
[ редактировать ]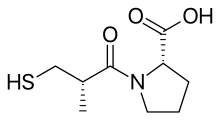
Более 2000 соединений были случайным образом протестированы в морских свинок тесте на подвздошной кишке , и было обнаружено, что сукцинил-L-пролин обладает свойствами специфического ингибитора АПФ. Он продемонстрировал ингибирующее действие ангиотензина I и брадикинина, не оказывая никакого влияния на ангиотензин II . Тогда исследователи начали поиск модели, которая могла бы объяснить ингибирование на основе специфического лекарственного взаимодействия соединений с активным центром АПФ. [ 5 ] Предыдущие исследования субстратов и ингибиторов АПФ показали, что это цинксодержащий металлопротеин и карбоксипептидаза, сходная с карбоксипептидазой А поджелудочной железы Однако АПФ высвобождает дипептиды, а не отдельные аминокислоты с С-конца пептидных . субстратов. И предполагалось, что и механизм их действия , и активный центр могут быть схожими. Положительно заряженный Arg 145 Считалось, что в активном центре связывается отрицательно заряженная С-концевая карбоксильная группа пептидного субстрата. Было также высказано предположение, что ACE связывается посредством водородной связи с концевой неразрывной пептидной связью субстрата. [ 3 ]
Но поскольку АПФ является дипептидкарбоксипептидазой, в отличие от карбоксипептидазы А, расстояние между катионным карбоксильным центром и атомом цинка должно быть больше, примерно на длину одного аминокислотного остатка. Пролин был выбран в качестве аминокислотной группы из-за его присутствия в качестве карбокси-концевого аминокислотного остатка в тепротиде и других ингибиторах АПФ, обнаруженных в змеиных ядах. Были протестированы еще одиннадцать аминокислот, но ни одна из них не оказалась более ингибирующей. Поэтому было предложено, чтобы производное сукциниловой аминокислоты было ингибитором АПФ, и было обнаружено, что сукцинил-L-пролин является таким ингибитором. [ 3 ] [ 5 ] [ 7 ]
Было также известно, что природа предпоследнего аминокислотного остатка пептидного субстрата АПФ влияет на связывание с ферментом. Ацильная группа карбоксиалканоиламинокислоты связывает ион цинка фермента и занимает в активном центре АПФ то же положение, что и предпоследняя. Следовательно, заместитель ацильной группы также может влиять на связывание с ферментом. Было обнаружено, что 2- метиловый заместитель с D-конфигурацией усиливает ингибирующую активность сукцинил-L-пролина примерно в 15 раз. Затем начались поиски лучшей группы, связывающей цинк. Замена сукцинилкарбоксильной группы азотсодержащими функциональными группами ( амином , амидом или гуанидином ) не усиливала ингибирующую активность. Однако прорыв в эффективности был достигнут за счет замены карбоксильной группы на сульфгидрильную функцию ( SH ), группу с большим сродством к иону цинка, связанному с ферментом. В результате был получен мощный ингибитор, который был в 1000 раз более мощным, чем сукцинил-L-пролин. [ 3 ] [ 7 ] меркаптоалканоилпроизводных . - Установлено, что оптимальная длина ацильной цепи для меркаптоалканоилпроизводных пролина равна 3-меркаптопропаноил-L-пролину, в 5 раз больше, чем у 2-меркаптоалканоилпроизводных, и в 50 раз больше, чем у 4 Таким образом, D-3-меркапто-2-метилпропаноил-L-пролин или каптоприл оказался наиболее мощным ингибитором. Позже исследователи сравнили несколько ингибиторов меркаптоацил-аминокислот и пришли к выводу, что связывание ингибитора с ферментом включает водородную связь между донорным сайтом фермента и кислородом амид-карбонила, как и предполагалось для субстратов. [ 3 ] [ 8 ]
Разработка лекарств других ингибиторов АПФ первого поколения
[ редактировать ]

Наиболее частые побочные эффекты каптоприла, кожная сыпь и потеря вкуса , такие же, как и при приеме меркаптосодержащего пеницилламина . Поэтому группа исследователей стремилась найти мощные селективные ингибиторы АПФ, которые не содержали бы меркапто (SH) функции и имели бы более слабую хелатирующую функцию. Они вернулись к работе с карбоксильными соединениями и начали работать с замещенными N -карбоксиметилдипептидами общей структуры (R-CHCOOH-A 1 -A 2 ). Согласно предыдущим исследованиям, они предположили, что циклические иминокислоты будут иметь хорошую эффективность, если их заменить на карбоксильном конце дипептида. Поэтому замена А 2 на пролин дала хорошие результаты. Они также отметили, что согласно специфичности фермента иминокислоты в положении рядом с карбоксильным концом не дают сильнодействующего соединения. Замещение R и A 1 групп гидрофобными и основными остатками позволит получить сильнодействующее соединение. Замена –NH в общей структуре привела к потере активности, что соответствует потребности фермента в –NH в соответствующем положении на субстратах. В результате были получены 2 активных ингибитора: Эналаприлат и Лизиноприл . Оба этих соединения содержат фенилаланин в положении R, который занимает бороздку S1 в ферменте. Результатом стали два новых мощных аналога трипептида с карбоксильной группой, координирующей цинк: эналаприлат и лизиноприл. [ 1 ] [ 9 ]
Обнаружение 2-х активных сайтов: C-домена и N-домена.
[ редактировать ]Большинство ингибиторов АПФ, представленных сегодня на рынке, неселективны по отношению к двум активным центрам АПФ, поскольку их связывание с ферментом основано главным образом на сильном фундаментальном взаимодействии между атомом цинка в ферменте и сильной хелатирующей группой ингибитора. Разрешение трехмерной структуры зародышевого АПФ, который имеет только один активный сайт, соответствующий С-домену соматического АПФ , предлагает структурную основу для структурного подхода к проектированию. Хотя N- и C-домен имеют сопоставимые скорости гидролиза АПФ in vitro , похоже, что in vivo С-домен в основном отвечает за регулирование артериального давления. Это указывает на то, что селективные ингибиторы С-домена могут иметь профиль, аналогичный профилю современных неселективных ингибиторов. Ангиотензин I в основном гидролизуется С-доменом in vivo, тогда как брадикинин гидролизуется обоими активными центрами. Таким образом, разработка селективного ингибитора С-домена позволит некоторую деградацию брадикинина за счет N-домена, и этой деградации может быть достаточно для предотвращения накопления избытка брадикинина, который наблюдается во время приступов ангионевротический отек . Избирательное ингибирование С-домена может привести к специализированному контролю артериального давления с меньшими вазодилататорами побочными эффектами, связанными с . С другой стороны, селективные ингибиторы N-домена открывают возможность открытия новых терапевтических областей. По-видимому, N-домен не играет большой роли в контроле артериального давления, но, по-видимому, является основным ферментом, метаболизирующим AcSDKP, естественный геморегуляторный гормон . [ 1 ] [ 10 ] [ 11 ]
Разработка лекарственного средства кето-АПФ и его кетометиленовых производных
[ редактировать ]Было обнаружено, что другие карбонилсодержащие группы, такие как кетоны, могут заменять амидную связь, связывающую Phe и Gly в ингибиторах АПФ. Кето-АПФ, впервые описанный в 1980 году, стал потенциальным ведущим соединением для ингибиторов АПФ, специфичных для С-домена. Кето-АПФ, трипептидный аналог Phe-Gly-Pro, содержит объемистые кольца P 1 и P 2 бензильные и, как было показано, ингибирует гидролиз ангиотензина I и брадикинина через C-домен. Синтез аналогов кето-АПФ с Trp или Phe в положении P 2 ' привел к заметному увеличению селективности C-домена , но введение алифатической группы P 2 обеспечило селективность N-домена. Ингибирующая активность может быть дополнительно усилена путем включения гидрофобного заместителя, такого как фенильная группа, в положение P 1 '. заместители P 1 ' с S- стереохимией обладают большей ингибирующей способностью, чем их R-аналоги. Также было показано, что [ 2 ] [ 8 ] [ 12 ] [ 13 ]
Кето-АПФ был использован в качестве основы для создания производных кетометилена. кетометилена Его аналоги содержат замену изостера в разрезаемой связи которая, как полагают, имитирует тетраэдра переходное состояние протеолитической , реакции в активном центре. Основное внимание было уделено простому трипептиду Phe-Ala-Pro, который в более ранних ферментных анализах показал ингибирующую активность. Замена аланина глицином дала трипептид с 1/14 ингибирующей активностью Phe-Ala-Pro. Бензоилированное производное Phe-Gly-Pro, Bz-Phe-Gly-Pro, было в два раза активнее. Чтобы уменьшить пептидную природу ингибиторов кетометилена, заместители P 1 ' и P 2 ' могут быть циклизованы с образованием лактама , где существует корреляция между ингибирующей активностью и размером кольца. В 2001 году было высказано предположение, что замена α на азот и создание 3-метилзамещенного аналога А58365А, пиридоновой кислоты, выделенной из ферментационного бульона бактерии Streptomyces с ингибирующей АПФ активностью , chromofuscus может влиять на уровень биологическая активность за счет стерического или гидрофобного эффекта и/или за счет предотвращения реакций на С3. также было замечено, Во время синтетической работы над A58365A что потенциальные предшественники чувствительны к окислению пятичленного кольца, и поэтому 3-метиловый аналог может быть более стабильным в этом отношении. [ 2 ] [ 14 ] [ 15 ]
Дизайн препарата силандиола
[ редактировать ]Тот факт, что углерод и силикон имеют схожие, но в то же время и разные характеристики, вызвал интерес к замене углерода силандиолом в качестве центральной группы, хелатирующей цинк. Силикон образует соединение диалкилсиландиола, которое достаточно затруднено, поэтому образование силоксанового полимера не происходит. Силандиолы более стабильны, чем углеродные диолы , поэтому ожидается, что они будут иметь более длительный период полураспада . Силандиолы также нейтральны при физиологическом pH (не ионизируются ). Четыре стереоизомера силандиола Phe-Ala сравнили с ингибиторами на основе кетонов, и выяснилось, что силандиол в четыре раза менее эффективен, чем кетоновый аналог. Это связано с тем, что силандиолы являются более слабыми хелаторами цинка по сравнению с кетонами. Замена силандиола на метилсиланогруппу приводила к незначительному ингибированию ферментов . Это подтверждает, что силандиоловая группа взаимодействует с АПФ как аналог переходного состояния и взаимодействие происходит аналогично кетону. [ 16 ] [ 17 ] Если бензильную группу силандиола заменить на изобутильную группу , получится более слабый ингибитор АПФ. Введение гидрофобного метилфенила дает немного большую эффективность, чем аналог с трет-бутильной группой в положении P 1 . Это говорит о том, что метилфенил лучше распознает S1, чем трет-бутильная группа. [ 2 ]
Фосфиновые пептиды
[ редактировать ]Фосфиновые пептиды представляют собой псевдопептиды, в которых связь фосфиновой кислоты (PO 2 -CH-) заменила пептидную связь в последовательности пептидного аналога. В некоторой степени химическая структура фосфиновых пептидов аналогична структуре промежуточных продуктов , образующихся при гидролизе пептидов протеолитическими ферментами. Была выдвинута гипотеза , что эти псевдопептиды имитируют структуру субстратов ферментов в их переходном состоянии, и кристаллография цинковых протеаз в комплексе с фосфиновыми пептидами подтверждает эту гипотезу. [ 10 ]
Дизайн препарата RXP 407
[ редактировать ]RXP 407 является первым фосфиновым пептидом, селективным по N-домену, и был обнаружен путем скрининга библиотек фосфиновых пептидов. До открытия RXP 407 долгое время утверждалось, что свободная C-концевая карбоксилатная группа в положении P 2 ' важна для эффективности ингибитора АПФ, поэтому можно предположить, что это отложило открытие селективных ингибиторов АПФ с N-доменом. . Когда был открыт RXP 407, исследователи изучили фосфиновые пептиды с тремя различными общими формулами, каждая из которых содержит 2 неопознанные аминокислоты, только одна из этих общих формул показала сильное ингибирование (Ac-Yaa-Pheψ(PO 2 -CH 2 )Ala-Yaa'- НХ 2 ). Были приготовлены пептидные смеси, заменяющие Yaa и Yaa' разными аминокислотами, пытаясь установить, существует ли мощный ингибитор, который мог бы ингибировать либо N-домен, либо C-домен фермента. В результате соединение Ac- Asp (L) -Pheψ(PO 2 -CH 2 ) (L) Ala-Ala-NH 2 активно ингибировало N-домен и получило название RXP 407. Взаимосвязь структура-функция показала, что карбоксамидная группа С-конца играла решающую роль в селективности N-домена АПФ. Кроме того, N-ацетильная группа и боковая цепь аспарагиновой кислоты в P 2- положение способствует селективности ингибитора в отношении N-домена. Эти особенности делают ингибитор недоступным для С-домена, но дают хорошую эффективность для N-домена, что приводит к разнице в ингибирующей способности активных центров на три порядка. Эти результаты также указывают на то, что N-домен обладает более широкой селективностью, чем C-домен. Еще одно различие между более старыми ингибиторами АПФ и RXP 407 заключается в молекулярном размере соединения. Старые ингибиторы АПФ в основном взаимодействовали с субсайтами S1 ' , S2 ' и S1, но RXP 407 взаимодействует дополнительно с субсайтом S2 . Это также важно для селективности ингибитора, поскольку боковая цепь аспарагиновой кислоты и N-ацетильная группа расположены в положении P 2 . [ 18 ]
Лекарственная конструкция RXPA 380
[ редактировать ]RXPA380 был первым ингибитором, обладающим высокой селективностью в отношении C-домена АПФ. Он имеет формулу Phe-Phe-Pro-Trp. [ 1 ] Разработка этого соединения была основана на исследованиях, которые показали, что некоторые пептиды, усиливающие брадикинин, проявляют селективность в отношении С-домена и все имеют в своей структуре несколько пролинов. Эти наблюдения побудили исследователей синтезировать фосфиновые пептиды, содержащие остаток пролина в положении P 1 ', и оценка этих соединений привела к открытию RXPA380. [ 19 ] Чтобы изучить роль остатков RXPA380, исследователи создали 7 аналогов RXPA380. Все полученные соединения были получены в виде смеси двух или четырех диастереоизомеров, но все они легко растворялись, и только одно из них было активным. Это согласуется с первоначальными исследованиями моделирования RXPA380, которые показали, что только один диастереомер может размещаться в активном центре зародышевого АПФ. Аналоги, в которых были заменены остатки псевдопролина или триптофана, показали меньшую селективность, чем RXPA380. Вероятно, это связано с тем, что эти два аналога обладают большей эффективностью в отношении N-домена, чем RXPA380. Замена обоих этих остатков дает большую эффективность, но не селективность. Это показывает, что остатки псевдопролина и триптофана хорошо приспосабливаются в C-домене, но не в N-домене. Еще два аналога, содержащие как псевдопролин, так и триптофан, но лишенные остатка псевдофенилаланина в положении P1, показали низкую активность в отношении N-домена, подобно RXPA380. Это подтверждает значительную роль этих двух остатков в селективности в отношении С-домена. Эти два аналога также обладают меньшей активностью в отношении C-домена, что показывает, что C-домен предпочитает псевдофенилаланиновую группу в P. 1 позиция. Моделирование комплекса RXPA380-ACE показало, что псевдопролиновый остаток ингибитора окружен аминокислотами, аналогичными аминокислотам N-домена, поэтому взаимодействия с доменом S 2 ' не могут быть ответственны за селективность RXPA380. Семь из 12 аминокислот, окружающих триптофан, одинаковы в C- и N-домене, самая большая разница заключается в том, что 2 объемные и гидрофобные аминокислоты в C-домене были заменены двумя меньшими и полярными аминокислотами в N-домене. Это указывает на то, что низкая активность RXPA380 в отношении N-домена обусловлена не тем, что полость S 2 ' не вмещает боковую цепь триптофана, а, скорее, тем, что отсутствуют важные взаимодействия между боковой цепью триптофана и аминокислотами C-домена. На основании близости между боковой цепью триптофана и Asp. 1029 возможна также водородная связь между карбоксилатом Asp. 1029 кольцо NH и индольное в C-домене, но в N-домене это взаимодействие гораздо слабее. [ 1 ]
Ссылки
[ редактировать ]- ^ Перейти обратно: а б с д и Ачарья, КР; Старрок, Эд; Риодан, Дж. К.; Элерс, MR (2003), «Пересмотр ACE: новая цель для структурного проектирования выемок», Nature Reviews Drug Discovery , 2 (11): 891–902, doi : 10.1038/nrd1227 , PMC 7097707 , PMID 14668810
- ^ Перейти обратно: а б с д Ределингхейс, П.; Нчинда, AT; Старрок, ED (2005), «Разработка домен-селективных ингибиторов ферментов», Annals of the New York Academy of Sciences , 1056 : 160–175, doi : 10.1196/annals.1352.035 , PMID 16387685 , S2CID 25407204
- ^ Перейти обратно: а б с д и ж Кушман, Д.В.; Чунг, HS; Сабо, EF; Ондетти, Массачусетс (1977). «Разработка мощных конкурентных ингибиторов ангиотензинпревращающего фермента. Карбоксиалканоил и меркаптоалканоил аминокислоты». Биохимия . 16 (25): 5484–5491. дои : 10.1021/bi00644a014 . ПМИД 200262 .
- ^ Кранц, Франция; Шварц, СЛ; Холленберг, Северная Каролина; Мур, Ти Джей; Длуги, Р.Г.; Уильямс, GH (1980), «Различия в реакции на ингибиторы пептидилдипептидгидролазы SQ 20 881 и SQ 14 225 при эссенциальной гипертензии с нормальным ренином», Hypertension , 2 (5): 604–609, doi : 10.1161/01.hyp.2.5. 604 , ПМИД 6158478
- ^ Перейти обратно: а б с Кушман, Д.В.; Ондетти, Массачусетс (1991), «История создания каптоприла и родственных ингибиторов ангиотензинпревращающего фермента», Hypertension , 17 (4): 589–592, doi : 10.1161/01.hyp.17.4.589 , PMID 2013486
- ^ Байерс, Л.Д.; Вулфенден, Р. (1973), «Связывание аналога побочного продукта бензилянтарной кислоты карбоксипептидазой А», Biochemistry , 12 (11): 2070–2078, doi : 10.1021/bi00735a008 , PMID 4735879
- ^ Перейти обратно: а б Ондетти, Массачусетс; Рубин, Б.; Кушман, Д.В. (1977), «Разработка специфических ингибиторов ангиотензинпревращающего фермента: новый класс перорально активных антигипертензивных агентов», Science , 196 (4288): 441–444, Bibcode : 1977Sci...196..441O , дои : 10.1126/science.191908 , ПМИД 191908
- ^ Перейти обратно: а б Кондон, Мэн; и др. (1982), «Ингибиторы ангиотензинпревращающего фермента: важность амидного карбонила меркаптоацил-аминокислот для образования водородных связей с ферментом», Journal of Medicinal Chemistry , 25 (3): 250–258, doi : 10.1021/jm00345a011 , PMID 6279843
- ^ Патчетт, А.А.; и др. (1980), «Новый класс ингибиторов ангиотензинпревращающего фермента», Nature , 288 (5788): 280–283, Bibcode : 1980Natur.288..280P , doi : 10.1038/288280a0 , PMID 6253826 , S2CID 4246377
- ^ Перейти обратно: а б Дайв, В.; и др. (2004), «Обзор: фосфиновые пептиды как ингибиторы металлопротеиназы цинка», Cellular and Molecular Life Sciences , 61 (16): 2010–2019, doi : 10.1007/s00018-004-4050-y , PMC 11138626 , PMID 15316651 , S2CID 3079 7568
- ^ Герогиадис, Д.; Гуниасс, П.; Коттон, Дж.; Йотакис, А.; Дайв, В. (2004), «Структурные детерминанты RXPA380, мощного и высокоселективного ингибитора C-домена ангиотензинпревращающего фермента», Biochemistry , 43 (25): 8048–8054, doi : 10.1021/bi049504q , PMID 15209500
- ^ Нчинда, AT; Чибале, К.; Ределингхейс, П.; Стиррок, Э.Д. (2006), «Синтез новых аналогов кето-АПФ в качестве домен-селективных ингибиторов ангиотензин-превращающего фермента», Bioorganic & Medicinal Chemistry Letters , 16 (17): 4612–4615, doi : 10.1016/j.bmcl. 2006.06.003 , ПМИД 16784850
- ^ Ределингхейс, П.; Нчинда, AT; Чибале, К.; Старрок, Э.Д. (2006), «Новые кетометиленовые ингибиторы ангиотензин-превращающего фермента (АПФ): ингибирование и молекулярное моделирование», Biological Chemistry , 387 (4): 461–466, doi : 10.1515/BC.2006.061 , PMID 16606345 , S2CID 30710745
- ^ Алмквист, Р.Г.; Чао, WR; Эллис, Мэн; Джонсон, HL (1980), «Синтез и биологическая активность кетомиленового аналога трипептидного ингибитора ангиотензинпревращающего фермента», Journal of Medical Chemistry , 23 (12): 1392–1398, doi : 10.1021/jm00186a020 , PMID 6256550
- ^ Клайв, DLJ; Ян, Х.; Леванчук, Э.З. (2001), «Синтез и активность in vitro неэпимеризуемого аналога ингибитора ангиотензинпревращающего фермента A58365A», Comptes Rendus de l'Académie des Sciences - Series IIC - Chemistry , 4 (6): 505– 512, дои : 10.1016/с1387-1609(01)01263-4
- ^ Ким, Дж.; Зибурт, С.М. (2004), «Силандиоловые пептидомиметики. Оценка четырех диастереомерных ингибиторов АПФ», Bioorganic & Medicinal Chemistry Letters , 14 (11): 2853–2856, doi : 10.1016/j.bmcl.2004.03.042 , PMID 15125946
- ^ Ким, Дж.; Хьюитт, Г.; Кэрролл, П.; Зибурт, С.М. (2005), «Силандиоловые ингибиторы ангиотензинпревращающего фермента. Синтез и оценка четырех диастереомеров аналогов дипептида Phe[Si]Ala», Journal of Organic Chemistry , 70 (15): 5781–5785, doi : 10.1021/ jo048121v , ПМИД 16018669
- ^ Дайв, В.; и др. (1999), «RXP 407, фосфиновый пептид, является мощным ингибитором ангиотензин I-превращающего фермента, способного различать два его активных центра», PNAS , 96 (8): 4330–4335, Bibcode : 1999PNAS...96.4330D , doi : 10.1073/pnas.96.8.4330 , PMC 16332 , PMID 10200262
- ^ Георгиадис, Д.; Бо, Ф.; Чарни, Б; Коттин, Дж; Йотакис, А; Дайв, В. (2003), «Роль двух активных участков соматического ангиотензинпревращающего фермента в расщеплении ангиотензина I и брадикинина: идеи селективных ингибиторов», Circulation Research , 93 (2): 148–154, doi : 10.1161 /01.RES.0000081593.33848.FC , PMID 12805239