Липосаркома
| Липосаркома | |
|---|---|
 | |
| Гистопатология липосаркомы, окраска H&E: [1] - | |
| Специальность | Дерматология , общая хирургия онкология |
| Симптомы | Шишка под кожей, боль, отек, дисфункция органов. |
Липосаркомы являются наиболее распространенным подтипом сарком мягких тканей , на их долю приходится не менее 20% всех сарком у взрослых. [2] Саркомы мягких тканей — редкие новообразования , имеющие более 150 различных гистологических подтипов или форм. Липосаркомы возникают из липобластов- ( предшественников адипоцитов т.е. жировых клеток) в жировой (т.е. жировой) ткани . Жировая ткань распределена по всему телу, включая такие участки, как глубокие и более поверхностные слои подкожных тканей , а также менее доступные хирургическим путем участки, такие как забрюшинное пространство (т.е. пространство за брюшной полостью ) и висцеральный жир внутри брюшной полости . [3]
имеют сходство с жировыми клетками . Все липосаркомы состоят, по крайней мере, из некоторых клеток, которые при гистопатологическом исследовании под микроскопом [4] Тем не менее, липосаркомы имеют несколько форм, основанных на различиях в их клинических проявлениях (например, возрасте, гендерных предпочтениях, локализации опухолей, признаках и симптомах ), степени тяжести (т. е. способности инвазировать местные ткани, рецидивировать после хирургического удаления и метастазировать в дистальные области). тканей), генетические аномалии , прогнозы и предпочтительные схемы лечения. Всемирная организация здравоохранения в 2020 году реклассифицировала липосаркомы на пять более или менее различных форм: 1) атипичная липоматозная опухоль/высокодифференцированная липосаркома (WD-LPS); 2) дедифференцированная липосаркома (ДД-ЛПС); 3) миксоидная липосаркома; 4) плеоморфная липосаркома; и 5) миксоидная плеоморфная липосаркома. [5] ( Плеоморфность указывает на наличие клеток, которые имеют аномальные и часто большие различия в размере и форме и/или размере и форме их ядер.)
В то время как формы липосаркомы классифицируются как агрессивные и злокачественные или, в случае атипичной липоматозной опухоли/высокодифференцированной липосаркомы, как относительно неагрессивные и доброкачественные, [6] все пять форм липосаркомы могут локально инфильтрироваться с повреждением близлежащих тканей и органов, возникать в хирургически недоступных участках, прилегающих к жизненно важным органам (например, забрюшинном пространстве). [7] ), рецидивируют после хирургического удаления и прогрессируют до опасных для жизни заболеваний. Исследования, проведенные на сегодняшний день, показывают, что все пять форм липосаркомы, хотя они обычно поддаются лечению, по крайней мере первоначально, путем хирургической резекции, часто лишь незначительно реагируют на используемые в настоящее время химиотерапии и лучевой терапии схемы . Липосаркомы требуют широкого спектра дальнейших исследований, чтобы определить их чувствительность к различным лучевой терапии , химиотерапии и более новым схемам лечения, которые используются индивидуально и в различных комбинациях, которые включают, где это возможно, хирургическое удаление. [6]
Этимология [ править ]
«жировая опухоль» (множественное число липомата), 1830 г., медицинская латынь, от греческого Lipos «жир» (сущ.), от корня PIE *leip- «прилипать, прилипать», также используется для образования слов, обозначающих «жир», + - ома.
1650-е годы, «мясистые наросты» (множественное число липосаркомата), медицинская латынь, от латинизированной формы греческого слова sarkoma «мясистое вещество» (Гален), от sarkoun «производить плоть, становиться мясистым», от sarx (родительный падеж sarkos) «плоть», +-ома.
Формы липосарком [ править ]

Липосаркомы обычно представляют собой крупные опухоли (> 10 см), но могут быть практически любого размера. Они встречаются в основном у взрослых, и только 0,7% случаев приходится на детей младше 16 лет. [5] У взрослых липосаркомы возникают преимущественно в среднем возрасте и после него. [8] В очень редких случаях, встречающихся у детей и подростков, диагностируют преимущественно миксоидную форму липосаркомы. [5]
Пять форм липосаркомы следует отличать не только друг от друга, но и от некоторых других опухолей мягких тканей. Этими другими опухолями наряду с некоторыми из их отличительных гистопатологических особенностей являются: 1) диспластические липомы (т.е. доброкачественные опухоли, которые имеют участки некроза тканей и неопластические жировые клетки различного размера, содержащие ядра различного размера/формы ; эти неопластические клетки, в отличие от большинства неопластических клеток) в липосаркомах не наблюдается сверхэкспрессии гена MDM2 ); [9] 2) атипичные веретеноклеточные липомы (т.е. доброкачественные опухоли с умеренно атипичными веретенообразными клетками в фиброзно-миксоидной строме, смешанной с вакуолизированными липобластами и адипоцитами вариабельного размера с атипичными ядрами; 3) плеоморфные липомы (т.е. доброкачественные опухоли, характеризующиеся гигантскими клетками) с перекрывающимися ядрами); [8] и 4) одиночные фиброзные опухоли (т.е. опухоли, до 22% из которых проявляют злокачественный характер, состоящие из клеток веретенообразной или овоидной формы внутри коллагеновой фоновой стромы, смешанной с кровеносными сосудами характерной коралловидной формы). [10] ). [5]
/высокодифференцированная Атипичная липоматозная опухоль липосаркома
Вместе атипичные липоматозные опухоли (АЛТ) и высокодифференцированные липосаркомы (ЛВЛ) составляют 40–45% всех липосарком. [11] Они редко, если вообще когда-либо, метастазируют и поэтому считаются доброкачественными или предраковыми опухолями. [12] [13] Однако они локально инвазивны и могут трансформироваться в более агрессивную и потенциально метастазирующую липосаркому, т.е. в дедифференцированную липосаркому. Кроме того, удаленная хирургическим путем атипичная липоматозная опухоль/высокодифференцированная липосаркома может рецидивировать в виде дедифференцированной липосаркомы. [6]
Презентация [ править ]

ALT и WDL считаются практически идентичными опухолями, за исключением того, что по определению ALT обозначают опухоли, которые развиваются в руках или ногах, тогда как WDL обозначают опухоли, которые развиваются в менее доступных для хирургического вмешательства местах, таких как глубокие, расположенные в центре мягкие ткани забрюшинного пространства , паратестикулярная область ( т.е. область в мошонке , включая яички , семенной канатик , оболочку яичка , придаток яичка и придаток яичка ), [6] полость рта и глазница . [12] [14] Эта терминология имеет прогностическое значение: менее 7% опухолей АЛТ преобразуются в дедифференцированные липосаркомы в течение среднего времени 7 лет, тогда как 17% опухолей WDL преобразуются в более злокачественную липосаркому в течение среднего времени 8 лет. [6] Опухоли ALT и WDL (далее называемые ALT/WDL) обычно появляются у людей среднего и старшего возраста в виде медленно увеличивающихся образований, которые имеют тенденцию быть больше и на более поздней стадии, когда расположены в глубоких тканях. [8] [11] Эти опухоли обычно безболезненны и, если расположены поверхностно, легко заметны; они также могут вызывать обширный отек (т.е. отек из-за местного скопления жидкости) в пораженных областях, таких как бедро (см. рисунок рядом), из-за их проникновения в кровеносные и/или лимфатические сосуды, дренирующие участок опухоли. Глубоко расположенные опухоли ALT/WDL могут протекать бессимптомно, но, в зависимости от их локализации, вызывают серьезные признаки и/или симптомы нарушения функции любого из различных органов, в которые они инфильтрируются. К этим органам относятся органы, расположенные близко или в забрюшинном пространстве (например, кишечник, почки и почечные мочеточники ); паратестикулярная область; средостение (например, трахея легких и крупные бронхи ); и голова (например, ретробульбарное пространство за глазным яблоком). [12] [14]
Патология [ править ]

Гистопатологически опухоли ALT/WDL делятся на адипоцитарно-липомоподобные, склерозирующие и воспалительные варианты, причем наиболее распространенными являются адипоцитоподобные/липомоподобные варианты. Адипоцитарные/липомоподобные опухоли ALT/WDL состоят из долек зрелых жировых клеток, по-разному пересекающихся с неровными фиброзными перегородками (см. соседнюю , окрашенную H&E микрофотографию ). Склерозирующие опухоли ALT/WDL, второй наиболее распространенный вариант, развиваются преимущественно в забрюшинной и паратестикулярной областях; он состоит из рассеянных атипичных стромальных клеток на фоне коллагеновой (т.е. коллагенсодержащей ) стромальной ткани . редкие вакуоли , содержащие липобласты Эту ткань заселяют . Воспалительные опухоли ALT/WDL являются самым редким вариантом. они чаще всего встречаются в забрюшинном пространстве и состоят из хронических воспалительных клеток, например, лимфоцитов и плазматических клеток, а также редких лимфатических фолликулов, разбросанных по тканям, содержащим жировые клетки . [14]
Генетика [ править ]
Неопластические клетки в опухолях ALT/WDL содержат одну или несколько дополнительных кольцевых малых сверхкомплектных маркерных хромосом (sSMC) или аномальную гигантскую маркерную хромосому (т. или более генетического материала другой хромосомы). Эти аномальные хромосомы содержат дополнительные копии длинного плеча хромосомы 12 (также называемого плечом q ) на участках с 13 по 15. Этот участок хромосомы 12 включает MDM2 протоонкоген (ген, потенциально вызывающий опухоли при сверхэкспрессии ), расположенный на участке хромосомы 12. 15 [15] и CDK4 (ген, сверхэкспрессия которого способствует развитию различных опухолей), расположенный в полосе 14.1. [16] [17] Амплификация или (т.е. увеличение количества копий гена без пропорционального увеличения числа других генов) этих двух генов является высокочувствительным и специфичным индикатором того, что липосаркома представляет собой либо ALT/WDL, либо дедифференцированную липосаркому, а не любую другую форму липосаркомы липомы . [17] Помимо генов MDM2 и CDK4 , эта область хромосомы 13–15 также содержит гены TSPAN31 и HMGA2 , сверхэкспрессия которых связана с различными опухолями и/или раком. Было высказано предположение, что один или несколько из этих сверхэкспрессируемых генов способствуют и/или способствуют развитию и/или прогрессированию опухолей ALT/WDL. [8]
Диагностика [ править ]
Диагноз опухолей АЛТ/ЛПВ ставится на основании особенностей их клинической картины, гистопатологии и генетических данных. В частности, обнаружение в опухолевых клетках ALT/WDL сверхэкспрессированного гена MDM2 или CDK4 или наличие либо специфической ALT/WDL-ассоциированной sSMC, либо гигантской маркерной хромосомы (как определено секвенированием ДНК следующего поколения , сравнительной геномной гибридизацией , [18] и/или узкоспециализированный цитогенетический анализ G-бэндинга [19] ) убедительно подтверждает диагноз АЛТ/ЛПВП или дедифференцированной липосаркомы. Клиническая картина и гистопатологические различия между двумя последними формами липосаркомы обычно помогают различить их. [8]
и прогноз Лечение
Опухоли ALT/WDL лечатся путем радикальной хирургической резекции с удалением всех неопластических тканей опухоли. Однако в 30–50% случаев эти опухоли локально рецидивируют. Рецидивы чаще всего возникают в опухолях, расположенных в менее доступных местах, например, в забрюшинном пространстве, средостении и семенном канатике. Эти менее поддающиеся хирургической оценке опухоли имеют тенденцию к неоднократному рецидивированию и в конечном итоге могут привести к смерти из-за их повреждающего воздействия на жизненно важные органы. Хотя опухоли ALT/WDL имеют очень небольшой потенциал к метастазированию , около 10% преобразуются в явно злокачественную и потенциально метастазирующую форму липосаркомы - дедифференцированную липосаркому. Среднее время этой злокачественной трансформации составляет около 7–9 лет. [8] Кроме того, хирургически удаленная АЛТ/ЛПВ может рецидивировать через разный интервал времени в виде дедифференцированной липосаркомы. [6] Крупное рандомизированное контролируемое исследование, сравнивающее лучевую терапию с последующим хирургическим вмешательством и только хирургическое вмешательство при опухолях АЛТ/ЛПВ, выявило небольшую разницу между двумя режимами. Небольшие исследования с использованием селективных ингибиторов белковых продуктов генов CDK4 или MDM2, вовлеченных в АЛТ/ЛПВП, показали в лучшем случае лишь умеренные эффекты. Дальнейшие исследования с использованием этих или совершенно новых схем лечения находятся в стадии изучения. [8] Обзорное исследование, проведенное в 2012 году, показало, что 5- и 10-летняя выживаемость людей с АЛТ/ЛПВП составляет 100% и 87% соответственно. [20]
Новые методы лечения
Новые методы лечения АЛТ/ЛПВП аналогичны тем, которые перечислены в разделе «Новые методы лечения» Дедифференцированной липосаркомы. [ нужна ссылка ]
липосаркома Дедифференцированная
Дедифференцированные липосаркомы представляют собой злокачественные опухоли, которые примерно в 10% случаев развиваются на фоне существующей атипичной липоматозной опухоли/опухоли высокодифференцированной липосаркомы (АЛТ/ВДЛ) или в месте, где опухоль АЛТ/ВПЛ была удалена хирургическим путем. У лиц с диагнозом этой опухоли de novo мог быть АЛТ/ЛПВ, который прогрессировал до дедифференцированной липосаркомы, но оставался незамеченным, поскольку развивался бессимптомно в сильно секвестрированном участке, таком как забрюшинное пространство или брюшная полость. Многие клинические и генетические особенности дедифференцированных опухолей липосаркомы аналогичны таковым, обнаруженным в опухолях ALT/WDL. [8]
Презентация [ править ]
Дедифференцированные липоосаркомы (ДДЛ) чаще всего встречаются у людей среднего и старшего возраста с пиком заболеваемости в возрасте от шестого до восьмого десятилетия. [8] Редко эти опухоли развиваются у детей и подростков. [5] Опухоли ДДЛ чаще всего возникают в забрюшинном пространстве, но, как и АЛТ/ЛВЛ, могут возникать в конечностях, паратестикулярной области, средостении, голове или шее. [8] Менее 1% всех ЛДЛ развиваются в поверхностных мягких тканях. [8] или глазницу. [21] При поступлении опухоли DDL обычно безболезненны, имеют большие размеры, могут медленно и прогрессивно увеличиваться в течение многих лет. [8] а на обычных рентгеновских снимках обнаруживаются участки отложения кальция (примером которых является рис. 1 в разделе «Гистопатология липосарком»). [22] [23] Реже у больных наблюдаются признаки и/или симптомы, связанные с поражением органа опухолью (например, боль в животе, вызванная закупоркой кишечника или обструкцией мочевыводящих путей, вызванная закупоркой уретры ). Очень редко у людей с ЛДЛ наблюдаются один или несколько признаков или симптомов хронического воспаления (см. симптомы B ) и/или один из эндокринных , неврологических , слизисто-кожных , гематологических или других тканевых паранеопластических синдромов . Признаки и симптомы хронического воспаления и различных паранеопластических синдромов вызваны секрецией опухолями цитокинов , гормонов , простагландинов и /или других агентов системного действия; они полностью исчезают после успешного лечения DDL. [8]
Патология [ править ]
Гистопатологический (которые представляют собой опухоли , вид опухолей DDL (см. рис. 2 в разделе «Гистопатология липосарком» ниже) широко варьируется, но чаще всего демонстрирует черты недифференцированных плеоморфных сарком густо заселенные клетками различного размера и формы, содержащими ядра вариабельного размера и формы ) или веретеноклеточные саркомы (представляющие собой опухоли, состоящие из веретенообразных клеток на соединительнотканном фоне). В разных частях опухолей DDL часто наблюдаются различия во внешнем виде фоновых соединительных тканей: эти ткани могут быть миксоидными (т.е. состоящими из прозрачного, похожего на слизь вещества, которое при окрашивании стандартным методом окрашивания H&E кажется более синим или фиолетовым, чем красный). цвет нормальных тканей) или миксоколлагеновый (т.е. высокое содержание коллагеновых волокон на миксоидном фоне) и в ~5% случаев имеют участки остеоида (см. рис. 1 в приведенном ниже разделе «Гистопатология липосарком») или хрящевого материала . Опухоли также демонстрируют большие различия в клеточном составе. Например, до 10% опухолей DDL имеют участки с гистопатологией ALT/WDL. [8] и в редких случаях DDL есть участки, содержащие менинготелиоподобные завитки плоских клеток. [24] [25]
Генетика [ править ]
Неопластические клетки как в DDL, так и в ALT/WDL несут сходные небольшие дополнительные маркерные хромосомы (sSMC) и/или гигантские маркерные хромосомы, которые содержат дополнительные части q-плеча хромосомы 12 в полосах с 13 по 15. Эта хромосомная область включает два гена, связанных с развитием опухоли. , МДМ2 [26] и гены CDK4 . [27] [17] Наличие дополнительных копий этих двух генов и/или их сверхпродуцируемых белковых продуктов является высокочувствительным и специфичным индикатором того, что липоматозная опухоль представляет собой ALT/WDL или DDL, а не какой-либо другой тип липоматозной опухоли. [12] [17] Предполагается, что сверхэкспрессия генов MDM2 и CDK и/или другого генетического материала в sSMC или гигантских маркерных хромосомах способствует развитию и/или прогрессированию опухолей DDL, а также ALT/WDL. [28] Другие гены в sMMC и гигантской маркерной хромосоме, которые также сверхэкспрессируются в неопластических клетках ALT/WDL и DDL, включают HMGA2 , CPM , YEATS4, [29] и ДДИТ3 . Однако по сравнению с неопластическими клетками ALT/WDL неопластические клетки DDL: 1) экспрессируют более высокие уровни генов в двух аномальных хромосомах; это может способствовать прогрессированию ALT/WDL в DDL; и 2) более высокие уровни генных продуктов на длинном плече хромосомы 1 на участке 32, на длинном плече хромосомы 6 на участке 33 и, примерно в 25% случаев, на коротком плече хромосомы 1 на участке 32.2, которое содержит Ген JUN (этот ген сверхэкспрессируется в DDL, но не в ALT/WDL). Поскольку JUN продукт гена , c-jun , ингибирует гибель клеток и способствует пролиферации клеток, его перепроизводство может способствовать прогрессированию ALT/WDL в DDL и/или злокачественному образованию неопластических клеток DDL. [8] Профилирование экспрессии генов (т.е. измерение экспрессии продуктов тысяч генов, вырабатываемых клетками, тканями или опухолями) показало, что дифференцировка адипоцитов и метаболические пути в АЛТ/ЛПВ активируются , в то время как пути пролиферации клеток и реакции на повреждение ДНК активируются. в ДДЛ. [6]
Диагностика [ править ]
Гистопатологические данные DDL часто недостаточно ясны для постановки точного диагноза. Однако диагноз DDL поддерживается у лиц: опухоли которых содержат ALT/WDL в смеси с гистологическими компонентами DDL; с историей наличия предшествующего ALT/WDL; [8] или у которых имеется забрюшинная липосаркома (ЛДЛ составляет ~57% всех забрюшинных липосарком). Опухоли DDL лишь в редких случаях (<1% случаев) проявляются как поверхностные опухоли кожи; [8] почти в 5 раз реже, чем ALT/WDL, встречаются в глазнице; [14] [21] и крайне редко встречаются у детей. [5] Обнаружение амплификации опухолевых клеток MDM2 является диагностическим золотым стандартом в отличии WDL от липом, диспластических липом, атипичных веретеноклеточных сарком, плеоморфных липом и одиночных фиброзных опухолей. [8] С другой стороны, обнаружение в опухолевых клетках сверхэкспрессированного гена CDK4 или наличие специфических ALT/WDL-ассоциированных sSMC или гигантской маркерной хромосомы убедительно подтверждают диагноз DDL или ALT/WDL. [18] [19] Клиническая картина, гистопатология и генные различия (например, сверхэкспрессия гена cJUN в опухолевых клетках сильно способствуют диагностике DDL по сравнению с ATL/WDL) между двумя последними формами липосаркомы обычно помогают различить их. [8]
и прогноз Лечение
Полная хирургическая резекция обычно является рекомендуемым лечением первой линии при локализованных опухолях DDL. [6] Однако новые исследования показывают, что пациенты с опухолями DDL, которые ограничены конечностью или туловищем и имеют прогнозируемую 10-летнюю общую выживаемость, связанную с опухолью, 51% или менее, имеют улучшенные результаты, когда к химиотерапии (например, доксорубицин плюс ифосфамид ) добавляется химиотерапия. их хирургические схемы. [30] Для этих локализованных форм ЛДЛ периоперационной лучевой терапии в соответствии с рекомендациями Национальной комплексной онкологической сети . также можно рассмотреть возможность [8]
Забрюшинная ДЛЛ является наиболее распространенной, хирургически недоступной и серьезной формой ДДЛ: частота рецидивов составляет 66%, а общая пятилетняя выживаемость составляет 54%. [31] Основным вариантом лечения забрюшинного ЛДЛ является хирургическая резекция. Клиническое исследование фазы III выявило небольшую разницу в результатах лучевой терапии с последующей хирургической резекцией по сравнению с только хирургической резекцией при лечении забрюшинного ЛДЛ. [6] В других клинических исследованиях III фазы пациентам с ЛДЛ с недоступными забрюшинными и/или метастатическими опухолями проводилась химиотерапия первой линии, сравнивающая доксорубицин с доксорубицином плюс ифосфамид или доксорубицин с гемцитабином плюс доцетаксел . Другие исследования также изучали ценность различных схем химиотерапии. Эти исследования часто обнаруживали небольшую разницу в общем времени выживаемости в своих сравнениях, но показали некоторые улучшения в выживаемости без прогрессирования и других клинических параметрах. На основании этих исследований рекомендуемой терапией первой линии при забрюшинных и других хирургически неопределяемых или метастатических опухолях DDL является лечение химиотерапией на основе антрациклинов или, в резистентных к опухоли или рецидивирующих случаях, химиотерапия эрибулином . Обзор, проведенный в 2020 году, показал, что медиана времени выживаемости для DDL низкой гистопатологической степени и высокой гистопатологической степени составляет 113 месяцев и 48 месяцев соответственно. [32] Необходимы дальнейшие исследования, чтобы предоставить доказательства эффективности лучевой терапии, химиотерапии и новых методов лечения всех разновидностей ЛДЛ. [33]
Новые методы лечения
Несколько новых схем терапии ЛПНП и более агрессивных или других проблемных случаев АЛТ/ЛПВ в настоящее время проходят клинические испытания . абемациклиба у В настоящее время проводится клиническое исследование фазы II по изучению пациентов с предварительно леченными или нелеченными ЛДЛ. Предварительный анализ показал, что этот ингибитор CDK4 и CDK6 продуктов генов , ферментов серин/треонин-специфической протеинкиназы, приводит к увеличению медианного времени выживаемости без прогрессирования, составляющего 30,4 недели. [6] Многоцентровое -контролируемое клиническое исследование фазы III рандомизированное двойное слепое плацебо абемациклиба находится в активной фазе и вскоре (как заявлено в июле 2021 г.) начнет набор 108 человек с распространенным, рецидивирующим и/или метастатическим ЛДЛ. Исследование спонсируется Альянсом Саркомы для исследований посредством сотрудничества. [34] в сотрудничестве с Eli Lilly and Company . [35] Рибоциклиб , также являющийся ингибитором генов CDK4 и CDK6 , в сочетании с ингибитором mTOR , эверолимусом проходит фазу II клинических испытаний на людях с поздней стадией DDL или лейомиосаркомой . [35] Регистрационное исследование III фазы (т.е. крупное подтверждающее исследование, призванное установить приемлемый профиль пользы/безопасности для получения одобрения регулирующих органов по точно определенному показанию) оценивает безопасность и эффективность миламетана по сравнению с трабектедином у пациентов с неоперабельными (т.е. считается, что резекция вызывает неприемлемую заболеваемость или смертность) или метастатический ЛДЛ, который прогрессировал на фоне 1 или более предшествующих системных терапий, включая по крайней мере 1 терапию на основе антрациклинов. Спонсор Rain Therapeutics Inc в настоящее время набирает 160 человек для участия в исследовании. [36] Еще одно клиническое исследование III фазы изучает ингибитор MDM2 миладеметан. [37] по сравнению с трабектедином , блокатором онкогенного фактора транскрипции FUS-CHOP , при MDM2 ALT/WDL и DDL. сверхэкспрессии [38] Миладеметан продемонстрировал контролируемую токсичность и некоторую активность, приводящую к стабилизации заболевания и/или нескольким частичным ответам на ЛДЛ. [6]
Миксоидная липосаркома [ править ]
Презентация [ править ]
Миксоидная липосаркома (MLS), которая включает тип липосаркомы, называемый круглоклеточной липосаркомой, [39] представляет ~30% всех липосарком. Пик заболеваемости приходится на четвертое и пятое десятилетия жизни, причем в большинстве исследований преобладают мужчины. Хотя MLS редко встречается у детей и подростков, он является наиболее распространенной формой липосаркомы, диагностируемой в этих возрастных группах. MLS обычно представляет собой большое (от 1 до 39 см; в среднем 12 см) подвижное, хорошо очерченное, безболезненное образование, которое развилось за период от 1 недели до 15 лет до постановки диагноза. Опухоли МЛС локализуются в глубоких мягких тканях бедер (65–80% случаев), голеней (10–15% случаев), забрюшинного пространства (8% случаев) и рук (5% случаев). Примерно в трети случаев эти опухоли метастазируют в другие участки мягких тканей (например, забрюшинное пространство, грудную клетку или другие конечности), кости скелета и/или легкие. У отдельных людей могут быть такие метастазы, особенно в костях; было рекомендовано, чтобы пациенты при поступлении проходили обследование на наличие метастазов в костях с помощью медицинской визуализации , включая рентген , компьютерную томографию и/или магнитно-резонансную томографию. . [40]
Патология [ править ]
Гистопатологический анализ MLS (см. рисунки 3 и 4 в разделе «Гистопатология липосарком» ниже) выявляет клетки, разбросанные по миксоидному матриксу (т.е. фон соединительной ткани, который выглядит более синим или фиолетовым, чем красный цвет нормальной соединительной ткани, когда эти ткани должным образом подготовлены, окрашены H&E и исследованы под микроскопом). Эти клетки представляют собой липобласты , некоторые из которых имеют форму кольца-печатки (форма позволяет предположить, что клетка может быть неопластической), овальной или круглой формы. [40] Опухоли MLS могут быть гиперклеточными и содержать сплошные пласты круглых клеток, которые составляют не менее 5% всех клеток, или с низкой клеточной структурой, заселенными клетками с мягкими ядрами и <5% круглых клеток на фоне изогнутых капилляров, напоминающих проволочную сетку. Опухоли, которые содержат не менее 5% круглых клеток, классифицируются как опухоли высокой степени злокачественности, а опухоли с <5% круглых клеток классифицируются как опухоли низкой степени злокачественности. [39] Опухоли MLS высокой степени злокачественности обычно имеют более агрессивное клиническое течение, чем опухоли MLS низкой степени злокачественности. [40]
Генетика [ править ]
Опухолевые клетки MLS фактически определяются по экспрессии FUS-DDIT3 слитого гена (также называемого химерным геном ), который встречается в >95% случаев, или слитого гена EWSR1-DDIT3 , который встречается в остальных <5% случаев. Слитый ген FUS -DDIT3 образуется в результате транслокации (называемой t(12:16)(q13:p11)) между сайтами DDIT3. гена [41] на участке 12 q-плеча 12-й хромосомы и на месте FUS гена [42] на участке 11 короткого плеча 16 хромосомы (также называемого p-плечом ). Известно, что слитый белок (также называемый химерным белком) этого химерного гена онкогена , FUS-DDIT3, останавливает созревание жировых клеток и способствует неоплазии. Слитый ген EWSR1-DDIT3 (названный t(12;22)(q13;q12)) возникает в результате транслокации EWSR1. гена [43] расположен в зоне 12.2 на q-плече 22-й хромосомы с геном DDIT2 . Продукт слитого белка гена EWSR1-DDIT3 , как и слитый белок FUS-DDIT3, способствует неоплазии. [44] Несмотря на эти родства слитых генов, необходимы дальнейшие исследования, чтобы определить их вклад в развитие и/или поддержание опухолей MLS. [6]
Диагностика [ править ]
Опухоли MLS низкой и средней степени злокачественности можно идентифицировать гистологически по классической морфологии характерной сетчатой сосудистой сети, разбросанной по миксоидной строме. Однако опухоли MLS высокой степени злокачественности бывает трудно отличить от других круглоклеточных новообразований, особенно от опухолей MLS высокой степени злокачественности, которые состоят из диффузно-клеточной и/или чисто круглоклеточной морфологии до такой степени, что скрывают этот классический сосудисто-миксоидный паттерн. Обнаружение реаранжировок гена DDIT3 с FUS или EWSR1 геном с помощью гибридизации in situ или иммуногистохимии или транскриптов слияния РНК этих генов с помощью полимеразной цепной реакции в реальном времени подтверждает диагноз как высокой степени, так и неоднозначных случаев низкой степени или опухоли MLS средней степени злокачественности. [44]
и прогноз Лечение
MLS обычно лечится хирургической резекцией, но может потребовать более радикальных вмешательств, например, может потребоваться ампутация конечности, когда -нервный пучок поврежден сосудисто конечности. Сообщается, что послеоперационный риск рецидива в течение 3 лет после операции составляет ~ 15%, если не вся опухоль удалена, и ~ 10%, когда удаление опухоли завершено. [40] Добавление лучевой терапии к хирургической резекции улучшило местный контроль опухолей MLS и было рекомендовано для лечения неоперабельного и рецидивирующего MLS. [45] Однако необходимы дальнейшие исследования, чтобы определить ценность лучевой терапии в лечении различных разновидностей МЛС. [40] Схемы химиотерапии с использованием ифосфамида , антрациклина, такого как даунорубицин , дакарбазин и/или трабектедин, оказались полезными: клиническое исследование III фазы показало, что время выживания без прогрессирования у пациентов с МЛС, получавших трабектедин или дакарбазин, составило 5,6 и 1,5 месяца соответственно. В 2015 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов одобрило использование трабектедина при неоперабельных и метастатических липосаркомах. [ нужна ссылка ]
В целом, 10-летняя выживаемость лиц с МЛС составила 77%, что значительно выше, чем при других формах липосаркомы. По сравнению с MLS низкого риска, MLS высокого риска (риск, определяемый содержанием круглых клеток опухоли и/или другими неблагоприятными прогностическими показателями) связан с повышенной частотой метастазирования и, следовательно, с более коротким временем выживания. Увеличение размера опухоли (≥ 10 см) тесно связано с MLS более высокой степени и, следовательно, с более коротким временем выживания. Другие факторы, которые были связаны с неблагоприятными исходами при MLS, включают наличие некроза опухоли, возраст> 45 лет, P53 , сверхэкспрессию гена [40] и мужской пол. [46] Круглоклеточная форма миксоидной липосаркомы также имеет относительно плохой прогноз: в различных ретроспективных обзорах миксоидная липосаркома обычно оказывалась низкодифференцированной и, следовательно, относительно чувствительной к химиотерапии, тогда как миксоидная липосаркома высокой степени злокачественности (т.е. круглоклеточная) имела более высокие показатели. метастазов, вели себя более агрессивно и плохо реагировали на химиотерапию. [40] Однако важно отметить, что почти все случаи миксоидных липосарком у педиатрических пациентов имели отличный прогноз. [45]
Новые методы лечения
Агонист PPAR -γ (т.е. активатор), эфатутазон, [47] был изучен в небольшом исследовании I фазы на людях с различными злокачественными новообразованиями на поздних стадиях. Препарат вызывал заметно стойкий ответ у человека с MLS, что позволяет предположить, что агонисты PPAR-γ могут быть полезны для лечения этого заболевания. [48] Клиническое исследование II стадии, проводимое в Италии, изучает эффекты трабектедина плюс пиоглитазона (еще одного агониста PPAR-γ) у людей со стабильными опухолями MLS. Исследование включает в себя два последовательных этапа. На первом этапе изучается реакция пациентов, получавших лечение только трабектедином в течение как минимум 4 циклов. Если будет достигнута стабилизация заболевания, на втором этапе будут изучены эффекты дальнейшего лечения первоначально ответивших пациентов комбинацией трабектедина и пиоглитазона. [49] Клинические испытания II стадии близятся к завершению для оценки эффективности сиролимуса (ингибитора MTOR ; сиролимус также известен как рапамицин) плюс циклофосфамида (химиотерапевтического препарата) при метастатическом или неоперабельном МЛС. [50] Клиническое исследование II фазы предполагает набор пациентов для оценки синтилимаба человека IgG4, ( моноклонального антитела направленного против белка 1 запрограммированной клеточной гибели, расположенного на поверхности клеток) в сочетании с двумя химиотерапевтическими препаратами, доксорубицином и ифосфамидом , в качестве лечения первой линии легкой степени тяжести. тканевые саркомы, включая MLS. [51]
Т-клетки были генетически сконструированы для воздействия на MAGE-A4 антиген , экспрессируемый на пептиде, содержащем HLA-A*02 MAGE-A4, расположенном на поверхности неопластических клеток в определенных типах опухолей. Эти сконструированные клетки (называемые клетками ADP-A2M4-T) атаковали и уничтожали различные культивированные раковые клетки человека, несущие этот антиген. [52] и в клиническом исследовании 1 стадии уменьшило различные типы солидных опухолей у пациентов, чьи опухоли содержали неопластические клетки, экспрессирующие этот антиген. [53] Клиническое исследование фазы II в настоящее время набирает людей для изучения эффективности и безопасности Т-клеток ADP-A2M4 (созданных из собственных Т-клеток реципиента) у HLA-A*02-положительных пациентов с метастатическим или неоперабельным, поздней стадией MSGE-4. -позитивные опухоли MLS. [54]
липосаркома Плеоморфная
Презентация [ править ]
Плеоморфные липосаркомы (ПЛС), на которые приходится от 5% до 10% всех случаев липосаркомы, [55] представляют собой быстрорастущие, обычно крупные (>5 см) и безболезненные, но высокозлокачественные опухоли адипоцитов. [56] Встречаются преимущественно у лиц старше 50 лет. [56] с преобладанием женщин. [45] Опухоли PLS редко встречаются у детей. [56] Опухоли PLS присутствуют в ноге или руке (65% случаев), забрюшинном пространстве или брюшной полости (15% случаев), [6] или в редких случаях стенка туловища, семенной канатик , [56] области головы и шеи, [57] грудная стенка, полость таза , легочная плевра , перикард и позвоночник. [55] Эти опухоли обычно локализуются в глубоких мягких тканях и только в 25% случаев локализуются в подкожных тканях. [56] Редкие случаи PLS наблюдались у людей с синдромами Ли-Фраумени или Мьюира-Торре , двумя наследственными генетическими нарушениями , которые предрасполагают больных к развитию различных типов рака. [45]
Патология [ править ]
Гистопатология опухолей PLS часто состоит из участков, напоминающих миксоидную липосаркому. [58] смешанные с участками, содержащими недифференцированные клетки. [56] различной формы Эти опухоли имеют выраженную гиперклеточную структуру и содержат по крайней мере несколько липобластов с плеоморфными ядрами. [58] Обычны участки некроза, гигантские клетки, некоторые из которых многоядерные и/или содержат поглощенные нейтрофилы иногда присутствуют , а в некоторых клетках можно увидеть капли гиалина , а также разбросаны внеклеточно по всей опухоли. [59] Недифференцированный компонент этих опухолей чаще всего состоит из веретенообразных клеток, при этом в 25% случаев обнаруживаются клетки с эпителиоидной морфологией клеток . Эти опухоли имеют по крайней мере несколько очагов с гистопатологией, сходной с гистиоцитомами миксофибросаркомы высокой степени злокачественности . [56] [58] [60] опухоль, ранее называвшаяся злокачественной миксоидной фиброзной гистиоцитомой. [61]
Генетика [ править ]
Неопластические клетки PLS содержат различные генные и хромосомные аномалии: TP53 ген удален или мутирован в 17–60% случаев; ген RB1 удален в 60% случаев; а ген нейрофибромина 1 теряется из-за инактивирующих мутаций в 8% случаев или, в более редких случаях, из-за делеции вокруг его местоположения в полосе 11.2 на длинном плече хромосомы 12. Эти клетки также могут демонстрировать увеличение генетического материала вокруг: полосы 12. –15 на коротком плече хромосомы 5; полоса 21 на коротком плече хромосомы 1; и полоса 22 на длинном плече хромосомы 7. Изменения числа копий гена, вызванные этими аномалиями, аналогичны тем, которые наблюдаются при миксофибросаркомном типе гистиоцитом . Роль(и) этих изменений в количестве копий гена в продвижении PLS не определена. Таким образом, PLS отличается от других липосарком тем, что его неопластические клетки имеют сложный геном без характерных геномных изменений или идентифицируемых генов, которые управляют заболеванием. Обнаружение изменений в экспрессии Гены TP53, RB1 и нейрофибромина 1 , а также другие, менее часто изменяемые гены при PLS (например, PIK3CA , тирозин-протеинкиназа SYK , PTK2B , EPHA5 и ERBB4 ) могут помочь поддержать, но не четко определить опухоль как Пожалуйста. [6] [56] Расширение теломер хромосомы заканчивается с помощью патологических механизмов, называемых альтернативным удлинением теломер, происходит в неопластических клетках примерно в 80% случаев PLS, но гораздо реже или не наблюдается в других четырех формах липосаркомы. [58]
Диагностика [ править ]
Диагноз PLS зависит от его проявления, гистопатологии и генетики. Гистопатология PLS часто очень напоминает миксофибросаркому, но отличается от этой опухоли содержанием плеоморфных липобластов. [58]
и прогноз Лечение
Радикальная хирургическая резекция является основным методом лечения ПЛС; это также важное паллиативное вмешательство для облегчения симптомов, вызванных сдавлением органов и тканей. Хирургическое вмешательство может потребовать удаления всего сдавленного органа, например почки или толстой кишки. Однако независимо от этой операции частота местных рецидивов очень высока. Не было доказано, что использование химиотерапии и/или лучевой терапии в сочетании с радикальным хирургическим вмешательством продлевает выживаемость и считается спорным вмешательством. [55] Национальная комплексная онкологическая сеть рекомендует пациентам с локализованным ПЛС высокого риска проводить полную хирургическую резекцию, если это возможно, в сочетании с лучевой терапией. Лиц с метастатическим заболеванием лечили химиотерапией (например, доксорубицин плюс ифосфамид или эрибулин ), аналогично схемам, используемым для дедифференцированной липосаркомы (см. раздел выше, посвященный лечению этого типа липосаркомы). [6] Около 20% опухолей PLS метастазируют в отдаленные места, наиболее распространенными из которых являются легкие (82% метастазов), печень (18% метастазов), кости или поджелудочная железа (18% метастазов). Сообщается, что выживаемость PLS через 1, 3 и 5 лет составляет 93%, 75% и 29% соответственно. Худший прогноз имеют опухоли, расположенные в центральном положении туловища, размером более 10 см, глубоко расположенные или содержащие участки некроза. [55]
липосаркома плеоморфная Миксоидная
Миксоидная плеоморфная липосаркома (первоначально называвшаяся плеоморфной миксоидной липосаркомой) [62] ) был впервые описан в большом исследовании липосарком в 2009 году. [63] Первоначально рассматриваемая как возможный вариант миксоидной липосаркомы с плеоморфными особенностями, Всемирная организация здравоохранения (2020) классифицировала ее как новую и особую форму липосаркомы. Эта классификация была основана на данных о том, что миксоидные плеоморфные липосаркомы, имея гистопатологические особенности, сходные с миксоидными липосаркомами, имели клинические и, что наиболее важно, критические генетические и молекулярные особенности, которые отличались от миксоидной, а также от трех других форм липосаркомы. [5]
Презентация [ править ]
Миксоидная плеоморфная липосаркома (МПЛ) — исключительно редкая и высокоагрессивная форма липосарком, развивающаяся у детей, подростков, [5] молодые люди, [6] и, в более недавнем исследовании, у людей старше 50 лет. [62] Опухоли MPL представляют собой глубокие образования мягких тканей, которые часто располагаются в средостении. [44] реже — конечности, голова и шея, брюшная полость или туловище. [6] По крайней мере, два случая MPL наблюдались у людей с синдромом Ли-Фраумени , наследственным генетическим заболеванием , которое предрасполагает людей к развитию различных видов рака. [58] [64] [65]
Патология [ править ]
По данным гистопатологического анализа, опухоли MPL состоят из участков, напоминающих обычную миксоидную липосаркому; эти области, составляющие 30–50% от общей площади опухоли, имеют обильный миксоидный матрикс, хорошо развитую капиллярную сосудистую сеть, мягкие клетки округлой и/или слегка веретенообразной формы, вакуолизированные липобласты и многоядерные клетки, имеющие форму небольших цветы. Однако эти области также содержат разбросанные высоко плеоморфные клетки, которые демонстрируют большую степень увеличения ядра и неравномерности, чем клетки миксоидных липосаркомных опухолей. Другие области опухолей MPL более клеточны и состоят из быстро растущих и высоко плеоморфных липобластов . [62]
Генетика [ править ]
Неопластические клетки при MPL не экспрессируют слитые гены FUS-DDIT3 или EWSR1-DDIT3 , которые экспрессируются неопластическими клетками в >95% или <5% случаев миксоидной фибросаркомы соответственно. [62] [6] Инактивация RB1 гена-супрессора опухоли вследствие его делеции или патологического подавления обнаруживается во всех случаях МПЛ. Неопластические клетки MPL также обычно имеют другие изменения в своих хромосомах. У них может наблюдаться аномальный прирост части генетического материала, обычно находящегося на хромосомах 1, 6, 7, 8, 19, 21 и/или X, и потери генетического материала, обычно находящегося на хромосомах 2, 3, 4, 5, 10. /или 22. Генетический материал, потерянный в полосе 14 на длинном плече хромосомы 13, включает не только ген RP1 , но также гены RCBTB2 , DLEU1 и , 11, 12, 13, 14, 15, 16, 17 и ITM2B . Из-за своей редкости и более позднего определения молекулярные характеристики и важность этих генетических аномалий еще не полностью определены. [56] Тем не менее, исследования показали, что потери в любом одном или нескольких генах RB1, RCBTB2, DLEU1 и ITM2B , но особенно в гене RP1 , могут способствовать развитию и/или прогрессированию MPL. [62]
Диагностика [ править ]
Диагноз MPL зависит от клинической картины опухоли, гистопатологического сходства с миксоидной липосаркомой и, что наиболее важно, отсутствия слитых генов FUS-DDIT3 sn EWSR1-DDIT3 в неопластических клетках. [62] [6]
и прогноз Лечение
В то время как людям с MPL лечили хирургическую резекцию для удаления опухолей, [64] [65] [6] [66] Обзор 2021 года показал, что не существует консенсусных рекомендаций по стандартам лечения MPL в отношении схем лучевой и химиотерапии (когда они используются отдельно или в сочетании с хирургическим вмешательством) для лечения этих опухолей. [6]
Гистопатология липосарком [ править ]
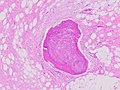 Рис. 1. Микрофотография костеобразования в опухоли липосаркомы.
Рис. 1. Микрофотография костеобразования в опухоли липосаркомы. Рис. 2. Микрофотография дедифференцированной опухоли липосаркомы.
Рис. 2. Микрофотография дедифференцированной опухоли липосаркомы. при меньшем увеличении. Рис. 3. Микрофотография миксоидной липосаркомы
при меньшем увеличении. Рис. 3. Микрофотография миксоидной липосаркомы в более высоком разрешении. Рис. 4. Микрофотография миксоидной липосаркомы
в более высоком разрешении. Рис. 4. Микрофотография миксоидной липосаркомы




Медицинская визуализация
Медицинская ультрасонография и магнитно-резонансная томография (МРТ) липосарком полезны и часто необходимы для определения их распространенности, хирургической доступности и связи с любыми наблюдаемыми органными дисфункциями. Поскольку ультразвуковое исследование обычно не позволяет отличить липосаркому от доброкачественной липомы, МРТ является первоначальным методом визуализации, позволяющим получить доказательства для проведения этого различия. [67]
При миксоидной липосаркоме на Т1-взвешенных МРТ-изображениях обнаруживаются массы с низкой интенсивностью сигнала и очаги с высокой интенсивностью сигнала. Масса демонстрирует высокую интенсивность сигнала на Т2-взвешенных изображениях. Это связано с тем, что он содержит преимущественно мукоидное вещество (обуславливает низкую интенсивность сигнала на Т1) и небольшое количество зрелого жира (обуславливает высокую интенсивность сигнала на Т1). [68] Образование четко очерченное, дольчатое, многокамерное или овальное по форме без какой-либо инфильтрации в окружающие структуры. [68]
![Рис. 5. Ультразвуковое исследование липосаркомы с высокоэхогенными областями, отраженными от липоматозного матрикса, и низкоэхогенными областями, отраженными от нелипоматозных областей.[69]](//upload.wikimedia.org/wikipedia/commons/thumb/a/a9/Scrotal_ultrasonography_of_liposarcoma.jpg/120px-Scrotal_ultrasonography_of_liposarcoma.jpg) Рис. 5. Ультрасонография липосаркомы с высокоэхогенными областями, отраженными от липоматозного матрикса, и низкоэхогенными областями, отраженными от нелипоматозных областей. [69]
Рис. 5. Ультрасонография липосаркомы с высокоэхогенными областями, отраженными от липоматозного матрикса, и низкоэхогенными областями, отраженными от нелипоматозных областей. [69]![Рис. 6. Ультразвуковое исследование липосаркомы, имитирующей липому. Это гомогенное высокоэхогенное образование имеет вид липомы.[69]](//upload.wikimedia.org/wikipedia/commons/thumb/3/34/Scrotal_ultrasonography_of_liposarcoma_mimicking_a_lipoma.jpg/120px-Scrotal_ultrasonography_of_liposarcoma_mimicking_a_lipoma.jpg) Рис. 6. Ультразвуковое исследование липосаркомы, имитирующей липому. Это гомогенное высокоэхогенное образование имеет вид липомы. [69]
Рис. 6. Ультразвуковое исследование липосаркомы, имитирующей липому. Это гомогенное высокоэхогенное образование имеет вид липомы. [69] Рис. 7. МРТ миксоидной липосаркомы высокой степени злокачественности в левой подмышечной области мужчины 40 лет, выделенной белым цветом, на этом горизонтальном срезе опухоли.
Рис. 7. МРТ миксоидной липосаркомы высокой степени злокачественности в левой подмышечной области мужчины 40 лет, выделенной белым цветом, на этом горизонтальном срезе опухоли.
![Рис. 5. Ультразвуковое исследование липосаркомы с высокоэхогенными областями, отраженными от липоматозного матрикса, и низкоэхогенными областями, отраженными от нелипоматозных областей.[69]](http://upload.wikimedia.org/wikipedia/commons/thumb/a/a9/Scrotal_ultrasonography_of_liposarcoma.jpg/120px-Scrotal_ultrasonography_of_liposarcoma.jpg)
![Рис. 6. Ультразвуковое исследование липосаркомы, имитирующей липому. Это гомогенное высокоэхогенное образование имеет вид липомы.[69]](http://upload.wikimedia.org/wikipedia/commons/thumb/3/34/Scrotal_ultrasonography_of_liposarcoma_mimicking_a_lipoma.jpg/120px-Scrotal_ultrasonography_of_liposarcoma_mimicking_a_lipoma.jpg)

и Общество культура
Известные случаи [ править ]
- Чад Браун (1961–2014), игрок в покер, умер от липосаркомы.
- Ричард Фейнман (1918–1988), физик-теоретик, умер после операции по лечению этой болезни.
- Роб Форд (1969–2016), бывший мэр Торонто и член городского совета Торонто, умер от плеоморфной липосаркомы.
- Хоки Гаджан (1959–2016), бывший бегун « Нью-Орлеан Сэйнтс» и радиокомментатор команды, умер от липосаркомы.
- Чарли Дэвис (1986 г.р.), бывший футболист Филадельфийского союза высшей лиги футбола, в 2016 году диагностировал липосаркому.
- Марк Стрэнд (1934–2014), бывший поэт-лауреат США и лауреат Пулитцеровской премии, умер от липосаркомы.
См. также [ править ]
- Липома
- Wendy Walk , некоммерческая организация, миссией которой является сбор средств и повышение осведомленности о саркомах, включая липосаркому.
Ссылки [ править ]
- ^ Сьюзан Поттервелд; Майкл Р. Клей. «Липосаркома» . Очерки патологии . Тема завершена: ноябрь 2017 г. Незначительные изменения: май 2023 г.
- ^ Не Л, Чен X, Гонг Дж, Чжан М, Сюй М, Чен Н, Чжоу Ц (декабрь 2020 г.). «Синхронная почечная дедифференцированная липосаркома и забрюшинная хорошо дифференцированная липосаркома: описание случая с обзором литературы». Международный журнал хирургической патологии . 29 (6): 667–671. дои : 10.1177/1066896920981682 . ПМИД 33355009 . S2CID 229688954 .
- ^ Деи Тос АП (август 2000 г.). «Липосаркома: новые сущности и развивающиеся концепции». Энн Диагн Патол . 4 (4): 252–66. дои : 10.1053/adpa.2000.8133 . ПМИД 10982304 .
- ^ Белл, Тереза (октябрь 2012 г.). «Что такое липосаркома?» . Инициатива по борьбе с саркомой Лидди Шрайвер . Проверено 22 апреля 2015 г.
- ↑ Перейти обратно: Перейти обратно: а б с д и ж г час Поррино Дж., Аль-Дасуки К., Иршаид Л., Ван А., Кани К., Хаймс А., Мэлони Э. (июнь 2021 г.). «Обновленные данные об опухолях мягких тканей у детей с обзором внешнего вида обычных МРТ. Часть 1: опухолеподобные поражения, адипоцитарные опухоли, фибробластические и миофибробластические опухоли, а также периваскулярные опухоли». Скелетная радиология . 51 (3): 477–504. дои : 10.1007/s00256-021-03836-2 . ПМИД 34191084 . S2CID 235678096 .
- ↑ Перейти обратно: Перейти обратно: а б с д и ж г час я дж к л м н тот п д р с т в Хэддокс К.Л., Ридель РФ (2021). «Последние достижения в понимании и лечении липосаркомы» . Обзоры факультетов . 10 :1. дои : 10.12703/r/10-1 . ПМЦ 7894267 . ПМИД 33659920 .
- ^ Нисио Дж (2011). «Вклад цитогенетики и молекулярной цитогенетики в диагностику адипоцитарных опухолей» . Журнал биомедицины и биотехнологии . 2011 : 524067. doi : 10.1155/2011/524067 . ПМК 3025394 . ПМИД 21274402 .
- ↑ Перейти обратно: Перейти обратно: а б с д и ж г час я дж к л м н тот п д р с т Туэй К (март 2019 г.). «Высокодифференцированная липосаркома и дедифференцированная липосаркома: обновленный обзор». Семинары по диагностической патологии . 36 (2): 112–121. дои : 10.1053/j.semdp.2019.02.006 . ПМИД 30852045 . S2CID 73725589 .
- ^ Михал М, Агайми А, Контрерас А.Л., Свайдлер М., Казаков Д.В., Штайнер П., Гроссманн П., Мартинек П., Хадравский Л., Михалова К., Свайдлер П., Шеп З., Михал М., Фетш Дж.Ф. (ноябрь 2018 г.). «Диспластическая липома: характерное атипичное липоматозное новообразование с анизоцитозом, фокальной ядерной атипией, сверхэкспрессией p53 и отсутствием амплификации гена MDM2 с помощью FISH; отчет о 66 случаях, демонстрирующих случайную мультифокальность и редкую связь с ретинобластомой». Американский журнал хирургической патологии . 42 (11): 1530–1540. дои : 10.1097/PAS.0000000000001129 . ПМИД 30001242 . S2CID 51616357 .
- ^ Мартин-Брото Дж., Мондаса-Эрнандес Дж.Л., Моура Д.С., Хинди Н. (июнь 2021 г.). «Комплексный обзор одиночной фиброзной опухоли: новые идеи для новых горизонтов» . Раки . 13 (12): 2913. doi : 10.3390/cancers13122913 . ПМЦ 8230482 . ПМИД 34200924 .
- ↑ Перейти обратно: Перейти обратно: а б Хамид М. (2007). «Патология и генетика адипоцитарных опухолей». Цитогенетические и геномные исследования . 118 (2–4): 138–47. дои : 10.1159/000108294 . ПМИД 18000364 . S2CID 27183708 .
- ↑ Перейти обратно: Перейти обратно: а б с д Пей Дж., Флидер Д.Б., Таларчек Дж.Н., Купер Х.С., Патчефски А.С., Вэй С. (март 2021 г.). «Клиническое применение хромосомного микроматричного анализа в диагностике липоматозных опухолей». Прикладная иммуногистохимия и молекулярная морфология . 29 (8): 592–598. дои : 10.1097/PAI.0000000000000923 . ПМИД 33734108 . S2CID 232299293 .
- ^ Усуда Д., Такэсима К., Санген Р., Накамура К., Хаяси К., Окамура Х., Каваи Ю., Касамаки Ю., Иинума Ю., Сайто Х., Канда Т., Урасима С. (октябрь 2018 г.). «Атипичная липоматозная опухоль круглой связки печени: описание случая и обзор литературы» . Всемирный журнал клинических случаев . 6 (12): 548–553. дои : 10.12998/wjcc.v6.i12.548 . ПМК 6212614 . ПМИД 30397612 .
- ↑ Перейти обратно: Перейти обратно: а б с д Кан Джи, Ким Х.Дж., Войно Т.Х., Юнг А.М., Мендоса П.Р., Гроссниклаус Х.Э. (2021). «Атипичная липоматозная опухоль/высокодифференцированная липосаркома орбиты: три случая и обзор литературы». Офтальмопластическая и реконструктивная хирургия . 37 (3С): С134–С140. дои : 10.1097/IOP.0000000000001804 . ПМИД 32991496 . S2CID 222143763 .
- ^ «Протоонкоген MDM2 MDM2 [Homo sapiens (Человек)] - Ген - NCBI» .
- ^ «Циклинзависимая киназа 4 CDK4 [Homo sapiens (Человек)] - Ген - NCBI» .
- ↑ Перейти обратно: Перейти обратно: а б с д Нисио Дж., Ивасаки Х., Сибата Т., Набешима К., Наито М. (апрель 2016 г.). «Дупликация сегмента хромосомы 12q13-15 в липоматозной опухоли с минимальной ядерной атипией: отчет о случае» . Письма об онкологии . 11 (4): 2875–2878. дои : 10.3892/ол.2016.4305 . ПМЦ 4812505 . ПМИД 27073568 .
- ↑ Перейти обратно: Перейти обратно: а б Слимани В., Джеллуль А., Аль-Рикаби А., Саллем А., Хасни Й., Чачиа С., Эрнез А., Чайеб А., Биби М., Лиер Т., Саад А., Мугу-Зерелли С. (июль 2020 г.). «Маленькие сверхкомплектные маркерные хромосомы (sSMC) и мужское бесплодие: характеристика пяти новых случаев, обзор литературы и перспективы» . Журнал вспомогательной репродукции и генетики . 37 (7): 1729–1736. дои : 10.1007/s10815-020-01811-9 . ПМЦ 7376793 . ПМИД 32399795 .
- ↑ Перейти обратно: Перейти обратно: а б Лу Ю, Лян Ю, Нин С, Дэн Г, Се Ю, Сун Дж, Цзо Н, Фэн С, Цинь Ю (2020). «Редкая частичная трисомия и тетрасомия 15q11-q13, связанная с задержкой развития и расстройством аутистического спектра» . Молекулярная цитогенетика . 13:21 . дои : 10.1186/s13039-020-00489-z . ПМЦ 7288499 . ПМИД 32536972 .
- ^ Гебхардт, М; Бюкер, П.Дж. (2004). «Липосаркома» . ЕСУН .
- ↑ Перейти обратно: Перейти обратно: а б Ямадзаки Д., Огихара Н., Хориучи Т. (июль 2020 г.). «Первичная орбитальная дедифференцированная липосаркома». Мировая нейрохирургия . 139 : 604–607. дои : 10.1016/j.wneu.2020.04.069 . ПМИД 32339743 . S2CID 216594976 .
- ^ Ямашита К, Кохаси К, Ямада Ю, Исии Т, Нисида Ю, Уракава Х, Ито И, Такахаси М, Иноуэ Т, Ито М, Охара Ю, Ода Ю, Тоёкуни С (апрель 2018 г.). «Остеогенная дифференцировка при дедифференцированной липосаркоме: исследование 36 случаев по сравнению со случаями без оссификации». Гистопатология . 72 (5): 729–738. дои : 10.1111/его.13421 . ПМИД 29076540 . S2CID 3829557 .
- ^ Чэнь Х.Г., Чжан К., Ву В.Б., У Ю.Х., Чжан Дж., Гу Л.Дж., Ли XJ (март 2020 г.). «Сочетание операции с брахитерапией 125I при рецидивирующей дедифференцированной липосаркоме средостения: описание случая и обзор литературы» . Всемирный журнал клинических случаев . 8 (5): 939–945. дои : 10.12998/wjcc.v8.i5.939 . ПМК 7062618 . ПМИД 32190631 .
- ^ Ли Дж., Фанг Л., Киллер Х.Э., Фламмер Дж., Мейер П., Нойцнер А. (июль 2013 г.). «Менинготелиальные клетки как часть защиты хозяина центральной нервной системы». Биология клетки . 105 (7): 304–15. дои : 10.1111/boc.201300013 . ПМИД 23634770 . S2CID 207094296 .
- ^ Усман Тарик М., Каяни Н., Моаттер Т., Дин НУ (октябрь 2020 г.). «Дедифференцированная липосаркома с менинготелиальными завитками: пять дополнительных случаев и обзор литературы». Международный журнал хирургической патологии . 28 (7): 749–758. дои : 10.1177/1066896920921950 . ПМИД 32419561 . S2CID 218680437 .
- ^ «Протоонкоген MDM2 MDM2 [Homo sapiens (Человек)] – Ген – NCBI» .
- ^ «Циклинзависимая киназа 4 CDK4 [Homo sapiens (Человек)] – Ген – NCBI» .
- ^ Маккиа Г, Севергнини М, Пургато С, Толомео Д, Кашаро Х, Чифола И, Л'Аббате А, Ловерро А, Палумбо О, Карелла М, Бьянкини Л, Перини Дж, Де Беллис Г, Мертенс Ф, Рокки М, Сторлацци КТ (март 2018 г.). «Скрытая геномная и транскриптомная пластичность гигантских маркерных хромосом при раке» . Генетика . 208 (3): 951–961. дои : 10.1534/genetics.117.300552 . ПМЦ 5844343 . ПМИД 29279323 .
- ^ «YEATS4 Домен YEATS, содержащий 4 [Homo sapiens (Человек)] – Ген – NCBI» .
- ^ Паскуали С., Пиццамильо С., Туати Н., Литьере С., Марро С., Каспер Б., Гелдерблом Х., Стаккиотти С., Джадсон И., Дей Тос А.П., Вердерио П., Казали П.Г., Уолл П.Дж., Грончи А. (март 2019 г.). «Влияние химиотерапии на выживаемость пациентов с саркомой мягких тканей конечностей и стенок туловища: пересмотр результатов рандомизированного исследования EORTC-STBSG 62931». Европейский журнал рака . 109 : 51–60. дои : 10.1016/j.ejca.2018.12.009 . ПМИД 30690293 . S2CID 59341549 .
- ^ Чэнь Дж, Ханг Ю, Гао Ц, Хуан Икс (2021). «Хирургическая диагностика и лечение первичной забрюшинной липосаркомы» . Границы в хирургии . 8 : 672669. дои : 10.3389/fsurg.2021.672669 . ПМК 8211986 . ПМИД 34150840 .
- ^ Амер К.М., Конгиуста Д.В., Томсон Дж.Э., Эльсамна С., Чаудри И., Боззо А., Амер Р., Сиракьюс Б., Герт М., Бибе К.С. (июль 2020 г.). «Эпидемиология и выживаемость липосаркомы и ее подтипов: анализ двойной базы данных» . Журнал клинической ортопедии и травматологии . 11 (Приложение 4): S479–S484. дои : 10.1016/j.jcot.2020.04.013 . ПМЦ 7394804 . ПМИД 32774015 .
- ^ Лам М.Б., Бальдини Э.Х., Рейджерс С.Дж., Хаас Р.Л., ДеЛэни Т.Ф. (июль 2021 г.). «Роль лучевой терапии при впервые диагностированной забрюшинной саркоме». Современные возможности лечения онкологии . 22 (9): 75. дои : 10.1007/s11864-021-00877-6 . hdl : 1887/3281923 . ПМИД 34213610 . S2CID 235701424 .
- ^ «Альянс Саркомы для исследований посредством сотрудничества» .
- ↑ Перейти обратно: Перейти обратно: а б «SARC041: Рандомизированное двойное слепое исследование фазы 3 абемациклиба по сравнению с плацебо у пациентов с поздней дедифференцированной липосаркомой» . 23 июля 2021 г.
- ^ «Рандомизированное многоцентровое исследование фазы 3 миладеметана по сравнению с трабектедином у пациентов с дедифференцированной липосаркомой» . 16 июля 2021 г.
- ^ «Миладеметан» .
- ^ «Рандомизированное многоцентровое исследование фазы 3 миладеметана по сравнению с трабектедином у пациентов с дедифференцированной липосаркомой» . 16 июля 2021 г.
- ↑ Перейти обратно: Перейти обратно: а б Зафар, Р.; Уилер, Ю. (2021). «Липосаркома» . СтатПерлс . СтатПерлз. PMID 30855853 .
- ↑ Перейти обратно: Перейти обратно: а б с д и ж г Муджтаба Б., Ван Ф., Тахер А., Аслам Р., Мадвелл Дж.Э., Нассар С. (2021). «Миксоидная липосаркома со скелетными метастазами: патофизиология и характеристики визуализации». Современные проблемы диагностической радиологии . 50 (1): 66–73. doi : 10.1067/j.cpradiol.2019.10.008 . ПМИД 31813645 . S2CID 208954696 .
- ^ «Транскрипт 3, индуцируемый повреждением ДНК DDIT3 [Homo sapiens (Человек)] – Ген – NCBI» .
- ^ «FUS FUS РНК-связывающий белок [Homo sapiens (Человек)] – Ген – NCBI» .
- ^ «РНК-связывающий белок 1 EWSR1 EWS [Homo sapiens (Человек)] – Ген – NCBI» .
- ↑ Перейти обратно: Перейти обратно: а б с Баранов Э., Блэк М.А., Флетчер К.Д., Чарвилл Г.В., Хорник Дж.Л. (июль 2021 г.). «Ядерная экспрессия DDIT3 отличает миксоидную липосаркому высокой степени злокачественности от других круглоклеточных сарком» . Современная патология . 34 (7): 1367–1372. дои : 10.1038/s41379-021-00782-1 . ПМЦ 8763641 . ПМИД 33731886 . S2CID 232247392 .
- ↑ Перейти обратно: Перейти обратно: а б с д Путра Дж., Аль-Ибрахими А. (март 2019 г.). «Адипоцитарные опухоли у детей: современный обзор». Семинары по диагностической патологии . 36 (2): 95–104. дои : 10.1053/j.semdp.2019.02.004 . ПМИД 30850231 . S2CID 73513220 .
- ^ Хейс Б., Холст-Берналь С., де Грааф М.А., Бриэр-де Брёйн И.Х., Родригес-Жирондо М., ван де Санде М.А., Вурер М., Макдоннелл Л.А., Бове Й.В. (сентябрь 2020 г.). «Молекулярные признаки прогрессирования опухоли при миксоидной липосаркоме, выявленные с помощью масс-спектрометрии N-гликанов» . Лабораторное исследование . 100 (9): 1252–1261. дои : 10.1038/s41374-020-0435-2 . ПМИД 32341520 . S2CID 216560584 .
- ^ «Эфатутазона дигидрохлорид» .
- ^ «Исследование фазы II гамма-агониста рецептора, активируемого пролифератором пероксисом, эфатутазона у пациентов с ранее леченной неоперабельной миксоидной липосаркомой» . 16 августа 2021 г.
- ^ «Исследование фазы II трабектедина в сочетании с агонистом PPARg пиоглитазоном у пациентов с круглоклеточными миксоидными липосаркомами или дедифференцированными липосаркомами G1 и G2 со стабильным заболеванием после монотерапии трабектедином. (TRABEPIO)» . 10 марта 2021 г.
- ^ «Фаза 2, одногрупповое, многоцентровое исследование по оценке эффективности комбинации сиролимуса и циклофосфамида при метастатической или неоперабельной миксоидной липосаркоме и хондросаркоме» . 4 июня 2021 г.
- ^ Ло, Чжиго (13 июля 2020 г.). «Одногрупповое, многоцентровое, исследование фазы II синтилимаба, доксорубицина и ифосфамида при лечении первой линии саркомы мягких тканей, включая недифференцированную плеоморфную саркому, синовиальную саркому, миксоидную липосаркому и дедифференцированную липосаркому» .
- ^ Сандерсон Дж.П., Кроули DJ, Видерманн Дж.Э., Куинн Л.Л., Кроссленд К.Л., Танбридж Х.М., Корнфорт ТВ, Барнс К.С., Ахмед Т., Хау К., Сэйни М., Эбботт Р.Дж., Андерсон В.Е., Тавано Б., Марото М., Джерри А.Б. (2020) . «Доклиническая оценка MAGE-A4-специфического Т-клеточного рецептора с повышенным сродством для адоптивной Т-клеточной терапии» . Онкоиммунология . 9 (1): 1682381. дои : 10.1080/2162402X.2019.1682381 . ПМК 6959444 . ПМИД 32002290 .
- ^ «Т-клетки, нацеленные на уменьшение опухолей MAGE-A4» . Открытие рака . 10 (8): ОФ2. Август 2020 г. doi : 10.1158/2159-8290.CD-NB2020-059 . ПМИД 32540953 .
- ^ «Открытое клиническое исследование фазы 2 с использованием Т-клеток ADP-A2M4 SPEAR™ у пациентов с распространенной синовиальной саркомой или миксоидной/круглоклеточной липосаркомой» . 18 июня 2021 г.
- ↑ Перейти обратно: Перейти обратно: а б с д Ван Л., Луо Р., Сюн З., Сюй Дж., Фан Д. (февраль 2018 г.). «Плеоморфная липосаркома: анализ 6 историй болезни и обзор литературы» . Лекарство . 97 (8): e9986. дои : 10.1097/MD.0000000000009986 . ПМК 5841962 . ПМИД 29465602 .
- ↑ Перейти обратно: Перейти обратно: а б с д и ж г час я Либбрехт С., Ван Дорпе Дж., Крейтенс Д. (март 2021 г.). «Быстро расширяющаяся группа опухолей мягких тканей с удаленным RB1: обновленный обзор» . Диагностика . 11 (3): 430. doi : 10.3390/diagnostics11030430 . ПМЦ 8000249 . ПМИД 33802620 .
- ^ Агарвал Дж., Кадакиа С., Агайми А., Огадзанов А., Хорсанди А., Чай Р.Л. (2017). «Плеоморфная липосаркома головы и шеи: представление двух случаев и обзор литературы». Американский журнал отоларингологии . 38 (4): 505–507. дои : 10.1016/j.amjoto.2017.04.012 . ПМИД 28528729 .
- ↑ Перейти обратно: Перейти обратно: а б с д и ж Демикко Э.Г. (март 2019 г.). «Молекулярные обновления адипоцитарных новообразований✰». Семинары по диагностической патологии . 36 (2): 85–94. дои : 10.1053/j.semdp.2019.02.003 . ПМИД 30857767 . S2CID 75135591 .
- ^ «Плеоморфная липосаркома» .
- ^ Огура К., Хосода Ф., Араи Ю., Накамура Х., Хама Н., Тотоки Ю., Ёсида А., Нагай М., Като М., Аракава Е., Мукаи В., Рокутан Х., Каваи А., Танака С., Сибата Т. (июль 2018 г.). «Комплексный генетический и эпигенетический анализ миксофибросаркомы» . Природные коммуникации . 9 (1): 2765. Бибкод : 2018NatCo...9.2765O . дои : 10.1038/s41467-018-03891-9 . ПМК 6050269 . ПМИД 30018380 .
- ^ Рашди I, Дауд Ф, Ханчел Ф, Арбауи И, Сомай М, Зубейди Х, Айди З, Бен Дхау Б, Деббиш А, Буссема Ф (декабрь 2020 г.). «Миксофибросаркома ноги: диагностическая проблема» . Отчеты о клинических случаях . 8 (12): 3333–3336. дои : 10.1002/ccr3.3414 . ПМЦ 7752563 . ПМИД 33363928 .
- ↑ Перейти обратно: Перейти обратно: а б с д и ж Крейтенс Д., Фольпе А.Л., Кельше С., Менцель Т., Фердинанде Л., ван Горп Дж.М., Ван дер Линден М., Раман Л., Ментен Б., Фритчи К., фон Даймлинг А., Ван Дорпе Дж., Флюке Ю. (июнь 2021 г.). «Миксоидная плеоморфная липосаркома - клинико-патологическое, иммуногистохимическое, молекулярно-генетическое и эпигенетическое исследование 12 случаев, предполагающее возможную связь с обычной плеоморфной липосаркомой» . Современная патология . 34 (11): 2043–2049. дои : 10.1038/s41379-021-00862-2 . ПМИД 34168281 . S2CID 235614580 .
- ^ Аладжо, Рита; Гроб, Шерил М.; Вайс, Шэрон В.; Бридж, Джулия А.; Иссаков, Жозефина; Оливейра, Андре М.; Фолпе, Эндрю Л. (май 2009 г.). «Липосаркомы у молодых пациентов: исследование 82 случаев, возникших у пациентов моложе 22 лет». Американский журнал хирургической патологии . 33 (5): 645–658. дои : 10.1097/PAS.0b013e3181963c9c . ПМИД 19194281 . S2CID 21863759 .
- ↑ Перейти обратно: Перейти обратно: а б Заре С.Ю., Лейво М., Фадаре О. (апрель 2020 г.). «Рецидивирующая плеоморфная миксоидная липосаркома у пациента с синдромом Ли-Фраумени». Международный журнал хирургической патологии . 28 (2): 225–228. дои : 10.1177/1066896919878804 . ПМИД 31559875 . S2CID 203568504 .
- ↑ Перейти обратно: Перейти обратно: а б Синклер Т.Дж., Торсон С.М., Альварес Э., Тан С., Спунт С.Л., Чао С.Д. (май 2017 г.). «Плеоморфная миксоидная липосаркома у подростка с синдромом Ли-Фраумени». Международная детская хирургия . 33 (5): 631–635. дои : 10.1007/s00383-017-4063-x . ПМИД 28160093 . S2CID 29704574 .
- ^ Пэн Р, Ли Н, Лан Т, Чен Х, Ду Т, Хэ Х, Чен М, Се Ю, Чжан Цз, Чжао В, Чжан Х (март 2021 г.). «Липосаркома у детей и молодых людей: клинико-патологическое и молекулярное исследование 23 случаев в одном из крупнейших учреждений Китая». Архив Вирхова . 479 (3): 537–549. дои : 10.1007/s00428-021-03076-8 . ПМИД 33738541 . S2CID 232273351 .
- ^ Рохит Шарма; Фрэнк Гайяр; и др. «Липома» . Радиопедия . Проверено 27 сентября 2018 г.
- ↑ Перейти обратно: Перейти обратно: а б Сун, Ми-Сук; Кан, Хеонг С.; Су, Джин С.; Ли, Юнг Х.; Пак, Чон М.; Ким, Джи Ю.; Ли, Хэ Г. (июль 2000 г.). «Миксоидная липосаркома: внешний вид при МРТ с гистологической корреляцией» . Радиографика . 20 (4): 1007–1019. doi : 10.1148/radiographics.20.4.g00jl021007 . ISSN 0271-5333 . ПМИД 10903690 .
- ↑ Перейти обратно: Перейти обратно: а б Первоначально контент скопирован с: Мак, Чи-Вай; Цзэн, Вэнь-Шэн (2012). «Сонография мошонки» . Сонография . дои : 10.5772/27586 . ISBN 978-953-307-947-9 . по лицензии CC-BY-3.0 .
| Базы данных органов управления : Национальные |
|---|