стр.53
| ТП53 | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | TP53, BCC7, LFS1, P53, TRP53, tumor protein p53, BMFS5, Genes, p53 | ||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 191170; MGI: 98834; HomoloGene: 460; GeneCards: TP53; OMA:TP53 - orthologs | ||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||





p53 , также известный как опухолевый белок P53 , клеточный опухолевый антиген p53 ( название UniProt ) или белок 53, связанный с трансформацией (TRP53), представляет собой регуляторный белок, который часто мутирует при раке человека. Белки p53 (первоначально считались и часто назывались одним белком) имеют решающее значение для позвоночных , поскольку они предотвращают образование рака . [ 5 ] Таким образом, р53 был описан как «хранитель генома » из-за его роли в сохранении стабильности путем предотвращения мутаций генома. [ 6 ] Отсюда TP53 [ примечание 1 ] классифицируется как ген-супрессор опухоли . [ 7 ] [ 8 ] [ 9 ] [ 10 ] [ 11 ]
Ген TP53 является наиболее часто мутирующим геном (>50%) при раке человека, что указывает на то, что ген TP53 играет решающую роль в предотвращении образования рака. [ 5 ] Ген TP53 кодирует белки, которые связываются с ДНК и регулируют экспрессию генов, предотвращая мутации генома. [ 12 ] Помимо полноразмерного белка, ген TP53 человека кодирует по меньшей мере 12 изоформ белка . [13]
Gene
[edit]In humans, the TP53 gene is located on the short arm of chromosome 17 (17p13.1).[7][8][9][10] The gene spans 20 kb, with a non-coding exon 1 and a very long first intron of 10 kb, overlapping the Hp53int1 gene. The coding sequence contains five regions showing a high degree of conservation in vertebrates, predominantly in exons 2, 5, 6, 7 and 8, but the sequences found in invertebrates show only distant resemblance to mammalian TP53.[14] TP53 orthologs[15] have been identified in most mammals for which complete genome data are available.
Human TP53 gene
[edit]In humans, a common polymorphism involves the substitution of an arginine for a proline at codon position 72 of exon 4. Many studies have investigated a genetic link between this variation and cancer susceptibility; however, the results have been controversial. For instance, a meta-analysis from 2009 failed to show a link for cervical cancer.[16] A 2011 study found that the TP53 proline mutation did have a profound effect on pancreatic cancer risk among males.[17] A study of Arab women found that proline homozygosity at TP53 codon 72 is associated with a decreased risk for breast cancer.[18] One study suggested that TP53 codon 72 polymorphisms, MDM2 SNP309, and A2164G may collectively be associated with non-oropharyngeal cancer susceptibility and that MDM2 SNP309 in combination with TP53 codon 72 may accelerate the development of non-oropharyngeal cancer in women.[19] A 2011 study found that TP53 codon 72 polymorphism was associated with an increased risk of lung cancer.[20]
Meta-analyses from 2011 found no significant associations between TP53 codon 72 polymorphisms and both colorectal cancer risk[21] and endometrial cancer risk.[22] A 2011 study of a Brazilian birth cohort found an association between the non-mutant arginine TP53 and individuals without a family history of cancer.[23] Another 2011 study found that the p53 homozygous (Pro/Pro) genotype was associated with a significantly increased risk for renal cell carcinoma.[24]
Function
[edit]DNA damage and repair
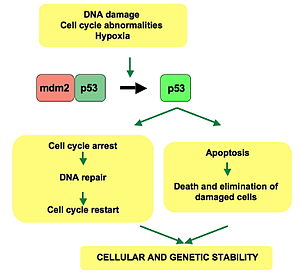
[edit]p53 plays a role in regulation or progression through the cell cycle, apoptosis, and genomic stability by means of several mechanisms:
- It can activate DNA repair proteins when DNA has sustained damage. Thus, it may be an important factor in aging.[25]
- It can arrest growth by holding the cell cycle at the G1/S regulation point on DNA damage recognition—if it holds the cell here for long enough, the DNA repair proteins will have time to fix the damage and the cell will be allowed to continue the cell cycle.
- It can initiate apoptosis (i.e., programmed cell death) if DNA damage proves to be irreparable.
- It is essential for the senescence response to short telomeres.
WAF1/CIP1 encodes for p21 and hundreds of other down-stream genes. p21 (WAF1) binds to the G1-S/CDK (CDK4/CDK6, CDK2, and CDK1) complexes (molecules important for the G1/S transition in the cell cycle) inhibiting their activity.
When p21(WAF1) is complexed with CDK2, the cell cannot continue to the next stage of cell division. A mutant p53 will no longer bind DNA in an effective way, and, as a consequence, the p21 protein will not be available to act as the "stop signal" for cell division.[26] Studies of human embryonic stem cells (hESCs) commonly describe the nonfunctional p53-p21 axis of the G1/S checkpoint pathway with subsequent relevance for cell cycle regulation and the DNA damage response (DDR). Importantly, p21 mRNA is clearly present and upregulated after the DDR in hESCs, but p21 protein is not detectable. In this cell type, p53 activates numerous microRNAs (like miR-302a, miR-302b, miR-302c, and miR-302d) that directly inhibit the p21 expression in hESCs.
The p21 protein binds directly to cyclin-CDK complexes that drive forward the cell cycle and inhibits their kinase activity, thereby causing cell cycle arrest to allow repair to take place. p21 can also mediate growth arrest associated with differentiation and a more permanent growth arrest associated with cellular senescence. The p21 gene contains several p53 response elements that mediate direct binding of the p53 protein, resulting in transcriptional activation of the gene encoding the p21 protein.
The p53 and RB1 pathways are linked via p14ARF, raising the possibility that the pathways may regulate each other.[27]
p53 expression can be stimulated by UV light, which also causes DNA damage. In this case, p53 can initiate events leading to tanning.[28][29]
Stem cells
[edit]Levels of p53 play an important role in the maintenance of stem cells throughout development and the rest of human life.
In human embryonic stem cells (hESCs)s, p53 is maintained at low inactive levels.[30] This is because activation of p53 leads to rapid differentiation of hESCs.[31] Studies have shown that knocking out p53 delays differentiation and that adding p53 causes spontaneous differentiation, showing how p53 promotes differentiation of hESCs and plays a key role in cell cycle as a differentiation regulator. When p53 becomes stabilized and activated in hESCs, it increases p21 to establish a longer G1. This typically leads to abolition of S-phase entry, which stops the cell cycle in G1, leading to differentiation. Work in mouse embryonic stem cells has recently shown however that the expression of P53 does not necessarily lead to differentiation.[32] p53 also activates miR-34a and miR-145, which then repress the hESCs pluripotency factors, further instigating differentiation.[30]
In adult stem cells, p53 regulation is important for maintenance of stemness in adult stem cell niches. Mechanical signals such as hypoxia affect levels of p53 in these niche cells through the hypoxia inducible factors, HIF-1α and HIF-2α. While HIF-1α stabilizes p53, HIF-2α suppresses it.[33] Suppression of p53 plays important roles in cancer stem cell phenotype, induced pluripotent stem cells and other stem cell roles and behaviors, such as blastema formation. Cells with decreased levels of p53 have been shown to reprogram into stem cells with a much greater efficiency than normal cells.[34][35] Papers suggest that the lack of cell cycle arrest and apoptosis gives more cells the chance to be reprogrammed. Decreased levels of p53 were also shown to be a crucial aspect of blastema formation in the legs of salamanders.[36] p53 regulation is very important in acting as a barrier between stem cells and a differentiated stem cell state, as well as a barrier between stem cells being functional and being cancerous.[37]
Other
[edit]
Apart from the cellular and molecular effects above, p53 has a tissue-level anticancer effect that works by inhibiting angiogenesis.[38] As tumors grow they need to recruit new blood vessels to supply them, and p53 inhibits that by (i) interfering with regulators of tumor hypoxia that also affect angiogenesis, such as HIF1 and HIF2, (ii) inhibiting the production of angiogenic promoting factors, and (iii) directly increasing the production of angiogenesis inhibitors, such as arresten.[39][40]
p53 by regulating Leukemia Inhibitory Factor has been shown to facilitate implantation in the mouse and possibly human reproduction.[41]
The immune response to infection also involves p53 and NF-κB. Checkpoint control of the cell cycle and of apoptosis by p53 is inhibited by some infections such as Mycoplasma bacteria,[42] raising the specter of oncogenic infection.
Regulation
[edit]p53 acts as a cellular stress sensor. It is normally kept at low levels by being constantly marked for degradation by the E3 ubiquitin ligase protein MDM2.[43] p53 is activated in response to myriad stressors – including DNA damage (induced by either UV, IR, or chemical agents such as hydrogen peroxide), oxidative stress,[44] osmotic shock, ribonucleotide depletion, viral lung infections[45] and deregulated oncogene expression. This activation is marked by two major events. First, the half-life of the p53 protein is increased drastically, leading to a quick accumulation of p53 in stressed cells. Second, a conformational change forces p53 to be activated as a transcription regulator in these cells. The critical event leading to the activation of p53 is the phosphorylation of its N-terminal domain. The N-terminal transcriptional activation domain contains a large number of phosphorylation sites and can be considered as the primary target for protein kinases transducing stress signals.
The protein kinases that are known to target this transcriptional activation domain of p53 can be roughly divided into two groups. A first group of protein kinases belongs to the MAPK family (JNK1-3, ERK1-2, p38 MAPK), which is known to respond to several types of stress, such as membrane damage, oxidative stress, osmotic shock, heat shock, etc. A second group of protein kinases (ATR, ATM, CHK1 and CHK2, DNA-PK, CAK, TP53RK) is implicated in the genome integrity checkpoint, a molecular cascade that detects and responds to several forms of DNA damage caused by genotoxic stress. Oncogenes also stimulate p53 activation, mediated by the protein p14ARF.
In unstressed cells, p53 levels are kept low through a continuous degradation of p53. A protein called Mdm2 (also called HDM2 in humans), binds to p53, preventing its action and transports it from the nucleus to the cytosol. Mdm2 also acts as an ubiquitin ligase and covalently attaches ubiquitin to p53 and thus marks p53 for degradation by the proteasome. However, ubiquitylation of p53 is reversible. On activation of p53, Mdm2 is also activated, setting up a feedback loop. p53 levels can show oscillations (or repeated pulses) in response to certain stresses, and these pulses can be important in determining whether the cells survive the stress, or die.[46]
MI-63 binds to MDM2, reactivating p53 in situations where p53's function has become inhibited.[47]
A ubiquitin specific protease, USP7 (or HAUSP), can cleave ubiquitin off p53, thereby protecting it from proteasome-dependent degradation via the ubiquitin ligase pathway. This is one means by which p53 is stabilized in response to oncogenic insults. USP42 has also been shown to deubiquitinate p53 and may be required for the ability of p53 to respond to stress.[48]
Recent research has shown that HAUSP is mainly localized in the nucleus, though a fraction of it can be found in the cytoplasm and mitochondria. Overexpression of HAUSP results in p53 stabilization. However, depletion of HAUSP does not result in a decrease in p53 levels but rather increases p53 levels due to the fact that HAUSP binds and deubiquitinates Mdm2. It has been shown that HAUSP is a better binding partner to Mdm2 than p53 in unstressed cells.
USP10, however, has been shown to be located in the cytoplasm in unstressed cells and deubiquitinates cytoplasmic p53, reversing Mdm2 ubiquitination. Following DNA damage, USP10 translocates to the nucleus and contributes to p53 stability. Also USP10 does not interact with Mdm2.[49]
Phosphorylation of the N-terminal end of p53 by the above-mentioned protein kinases disrupts Mdm2-binding. Other proteins, such as Pin1, are then recruited to p53 and induce a conformational change in p53, which prevents Mdm2-binding even more. Phosphorylation also allows for binding of transcriptional coactivators, like p300 and PCAF, which then acetylate the C-terminal end of p53, exposing the DNA binding domain of p53, allowing it to activate or repress specific genes. Deacetylase enzymes, such as Sirt1 and Sirt7, can deacetylate p53, leading to an inhibition of apoptosis.[50] Some oncogenes can also stimulate the transcription of proteins that bind to MDM2 and inhibit its activity.
Epigenetic marks like histone methylation can also regulate p53, for example, p53 interacts directly with a repressive Trim24 cofactor that binds histones in regions of the genome that are epigenetically repressed[51]. Trim24 prevents p53 from activating its targets, but only in these regions, effectively giving p53 the ability to 'read out' the histone profile at key target genes and act in a gene-specific manner.
Role in disease
[edit]
If the TP53 gene is damaged, tumor suppression is severely compromised. People who inherit only one functional copy of the TP53 gene will most likely develop tumors in early adulthood, a disorder known as Li–Fraumeni syndrome.
The TP53 gene can also be modified by mutagens (chemicals, radiation, or viruses), increasing the likelihood for uncontrolled cell division. More than 50 percent of human tumors contain a mutation or deletion of the TP53 gene.[52] Loss of p53 creates genomic instability that most often results in an aneuploidy phenotype.[53]
Increasing the amount of p53 may seem a solution for treatment of tumors or prevention of their spreading. This, however, is not a usable method of treatment, since it can cause premature aging.[54] Restoring endogenous normal p53 function holds some promise. Research has shown that this restoration can lead to regression of certain cancer cells without damaging other cells in the process. The ways by which tumor regression occurs depends mainly on the tumor type. For example, restoration of endogenous p53 function in lymphomas may induce apoptosis, while cell growth may be reduced to normal levels. Thus, pharmacological reactivation of p53 presents itself as a viable cancer treatment option.[55][56] The first commercial gene therapy, Gendicine, was approved in China in 2003 for the treatment of head and neck squamous cell carcinoma. It delivers a functional copy of the p53 gene using an engineered adenovirus.[57]
Certain pathogens can also affect the p53 protein that the TP53 gene expresses. One such example, human papillomavirus (HPV), encodes a protein, E6, which binds to the p53 protein and inactivates it. This mechanism, in synergy with the inactivation of the cell cycle regulator pRb by the HPV protein E7, allows for repeated cell division manifested clinically as warts. Certain HPV types, in particular types 16 and 18, can also lead to progression from a benign wart to low or high-grade cervical dysplasia, which are reversible forms of precancerous lesions. Persistent infection of the cervix over the years can cause irreversible changes leading to carcinoma in situ and eventually invasive cervical cancer. This results from the effects of HPV genes, particularly those encoding E6 and E7, which are the two viral oncoproteins that are preferentially retained and expressed in cervical cancers by integration of the viral DNA into the host genome.[58]
The p53 protein is continually produced and degraded in cells of healthy people, resulting in damped oscillation (see a stochastic model of this process in [59]). The degradation of the p53 protein is associated with binding of MDM2. In a negative feedback loop, MDM2 itself is induced by the p53 protein. Mutant p53 proteins often fail to induce MDM2, causing p53 to accumulate at very high levels. Moreover, the mutant p53 protein itself can inhibit normal p53 protein levels. In some cases, single missense mutations in p53 have been shown to disrupt p53 stability and function.[60]
 This image shows different patterns of p53 expression in endometrial cancers on chromogenic immunohistochemistry, whereof all except wild-type are variably termed abnormal/aberrant/mutation-type and are strongly predictive of an underlying TP53 mutation:[61]
|

Suppression of p53 in human breast cancer cells is shown to lead to increased CXCR5 chemokine receptor gene expression and activated cell migration in response to chemokine CXCL13.[64]
One study found that p53 and Myc proteins were key to the survival of Chronic Myeloid Leukaemia (CML) cells. Targeting p53 and Myc proteins with drugs gave positive results on mice with CML.[65][66]
Experimental analysis of p53 mutations
[edit]Most p53 mutations are detected by DNA sequencing. However, it is known that single missense mutations can have a large spectrum from rather mild to very severe functional effects.[60]
The large spectrum of cancer phenotypes due to mutations in the TP53 gene is also supported by the fact that different isoforms of p53 proteins have different cellular mechanisms for prevention against cancer. Mutations in TP53 can give rise to different isoforms, preventing their overall functionality in different cellular mechanisms and thereby extending the cancer phenotype from mild to severe. Recent studies show that p53 isoforms are differentially expressed in different human tissues, and the loss-of-function or gain-of-function mutations within the isoforms can cause tissue-specific cancer or provide cancer stem cell potential in different tissues.[11][67][68][69] TP53 mutation also hits energy metabolism and increases glycolysis in breast cancer cells.[70]
The dynamics of p53 proteins, along with its antagonist Mdm2, indicate that the levels of p53, in units of concentration, oscillate as a function of time. This "damped" oscillation is both clinically documented [71] and mathematically modelled.[72][73] Mathematical models also indicate that the p53 concentration oscillates much faster once teratogens, such as double-stranded breaks (DSB) or UV radiation, are introduced to the system. This supports and models the current understanding of p53 dynamics, where DNA damage induces p53 activation (see p53 regulation for more information). Current models can also be useful for modelling the mutations in p53 isoforms and their effects on p53 oscillation, thereby promoting de novo tissue-specific pharmacological drug discovery.
Discovery
[edit]p53 was identified in 1979 by Lionel Crawford, David P. Lane, Arnold Levine, and Lloyd Old, working at Imperial Cancer Research Fund (UK) Princeton University/UMDNJ (Cancer Institute of New Jersey), and Memorial Sloan Kettering Cancer Center, respectively. It had been hypothesized to exist before as the target of the SV40 virus, a strain that induced development of tumors. The name p53 was given in 1979 describing the apparent molecular mass.
The TP53 gene from the mouse was first cloned by Peter Chumakov of The Academy of Sciences of the USSR in 1982,[74] and independently in 1983 by Moshe Oren in collaboration with David Givol (Weizmann Institute of Science).[75][76] The human TP53 gene was cloned in 1984[7] and the full length clone in 1985.[77]
It was initially presumed to be an oncogene due to the use of mutated cDNA following purification of tumor cell mRNA. Its role as a tumor suppressor gene was revealed in 1989 by Bert Vogelstein at the Johns Hopkins School of Medicine and Arnold Levine at Princeton University.[78][79] p53 went on to be identified as a transcription factor by Guillermina Lozano working at MD Anderson Cancer Center.[80]
Warren Maltzman, of the Waksman Institute of Rutgers University first demonstrated that TP53 was responsive to DNA damage in the form of ultraviolet radiation.[81] In a series of publications in 1991–92, Michael Kastan of Johns Hopkins University, reported that TP53 was a critical part of a signal transduction pathway that helped cells respond to DNA damage.[82]
In 1993, p53 was voted molecule of the year by Science magazine.[83]
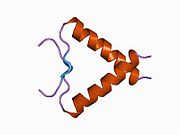
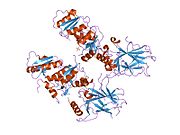
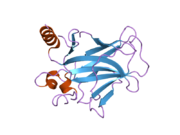
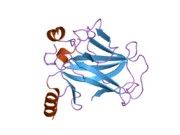
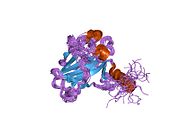
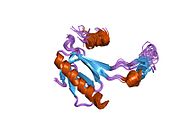
Structure
[edit]

p53 has seven domains:
- an acidic N-terminus transcription-activation domain (TAD), also known as activation domain 1 (AD1), which activates transcription factors. The N-terminus contains two complementary transcriptional activation domains, with a major one at residues 1–42 and a minor one at residues 55–75, specifically involved in the regulation of several pro-apoptotic genes.[84]
- activation domain 2 (AD2) important for apoptotic activity: residues 43–63.
- proline rich domain important for the apoptotic activity of p53 by nuclear exportation via MAPK: residues 64–92.
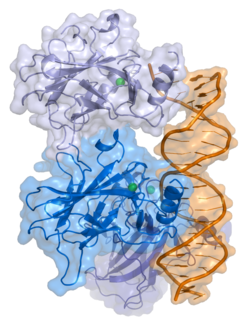
- central DNA-binding core domain (DBD). Contains one zinc atom and several arginine amino acids: residues 102–292. This region is responsible for binding the p53 co-repressor LMO3.[85]
- Nuclear Localization Signaling (NLS) domain, residues 316–325.
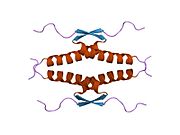
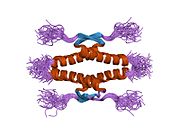
- homo-oligomerisation domain (OD): residues 307–355. Tetramerization is essential for the activity of p53 in vivo.
- C-terminal involved in downregulation of DNA binding of the central domain: residues 356–393.[86]
Mutations that deactivate p53 in cancer usually occur in the DBD. Most of these mutations destroy the ability of the protein to bind to its target DNA sequences, and thus prevents transcriptional activation of these genes. As such, mutations in the DBD are recessive loss-of-function mutations. Molecules of p53 with mutations in the OD dimerise with wild-type p53, and prevent them from activating transcription. Therefore, OD mutations have a dominant negative effect on the function of p53.
Wild-type p53 is a labile protein, comprising folded and unstructured regions that function in a synergistic manner.[87]
SDS-PAGE analysis indicates that p53 is a 53-kilodalton (kDa) protein. However, the actual mass of the full-length p53 protein (p53α) based on the sum of masses of the amino acid residues is only 43.7 kDa. This difference is due to the high number of proline residues in the protein, which slow its migration on SDS-PAGE, thus making it appear heavier than it actually is.[88]
Isoforms
[edit]As with 95% of human genes, TP53 encodes more than one protein. All these p53 proteins are called the p53 isoforms.[5] These proteins range in size from 3.5 to 43.7 kDa. Several isoforms were discovered in 2005, and so far 12 human p53 isoforms have been identified (p53α, p53β, p53γ, ∆40p53α, ∆40p53β, ∆40p53γ, ∆133p53α, ∆133p53β, ∆133p53γ, ∆160p53α, ∆160p53β, ∆160p53γ). Furthermore, p53 isoforms are expressed in a tissue dependent manner and p53α is never expressed alone.[11]
The full length p53 isoform proteins can be subdivided into different protein domains. Starting from the N-terminus, there are first the amino-terminal transcription-activation domains (TAD 1, TAD 2), which are needed to induce a subset of p53 target genes. This domain is followed by the proline rich domain (PXXP), whereby the motif PXXP is repeated (P is a proline and X can be any amino acid). It is required among others for p53 mediated apoptosis.[89] Some isoforms lack the proline rich domain, such as Δ133p53β,γ and Δ160p53α,β,γ; hence some isoforms of p53 are not mediating apoptosis, emphasizing the diversifying roles of the TP53 gene.[67] Afterwards there is the DNA binding domain (DBD), which enables the proteins to sequence specific binding. The C-terminus domain completes the protein. It includes the nuclear localization signal (NLS), the nuclear export signal (NES) and the oligomerisation domain (OD). The NLS and NES are responsible for the subcellular regulation of p53. Through the OD, p53 can form a tetramer and then bind to DNA. Among the isoforms, some domains can be missing, but all of them share most of the highly conserved DNA-binding domain.
The isoforms are formed by different mechanisms. The beta and the gamma isoforms are generated by multiple splicing of intron 9, which leads to a different C-terminus. Furthermore, the usage of an internal promoter in intron 4 causes the ∆133 and ∆160 isoforms, which lack the TAD domain and a part of the DBD. Moreover, alternative initiation of translation at codon 40 or 160 bear the ∆40p53 and ∆160p53 isoforms.[11]
Due to the isoformic nature of p53 proteins, there have been several sources of evidence showing that mutations within the TP53 gene giving rise to mutated isoforms are causative agents of various cancer phenotypes, from mild to severe, due to single mutation in the TP53 gene (refer to section Experimental analysis of p53 mutations for more details).
Interactions
[edit]p53 has been shown to interact with:
- AIMP2,[90]
- ANKRD2,[91]
- APTX,[92]
- ATM,[93][94][95][96][97]
- ATR,[93][94]
- ATF3,[98][99]
- AURKA,[100]
- BAK1,[101]
- BARD1,[102]
- BLM,[103][104][105][106]
- BRCA1,[102][107][108][109][110]
- BRCA2,[102][111]
- BRCC3,[102]
- BRE,[102]
- CEBPZ,[112]
- CDC14A,[113]
- Cdk1,[114][115]
- CFLAR,[116]
- CHEK1,[103][117][118]
- CCNG1,[119]
- CREBBP,[120][121][122]
- CREB1,[122]
- Cyclin H,[123]
- CDK7,[123][124]
- DNA-PKcs,[94][117][125]
- E4F1,[126][127]
- EFEMP2,[128]
- EIF2AK2,[129]
- ELL,[130]
- EP300,[121][131][132][133]
- ERCC6,[134][135]
- GNL3,[136]
- GPS2,[137]
- GSK3B,[138]
- HSP90AA1,[139][140][141]
- HIF1A,[142][143][144][145]
- HIPK1,[146]
- HIPK2,[147][148]
- ХМГБ1 , [ 149 ] [ 150 ]
- HSPA9 , [ 151 ]
- Хантингтин , [ 152 ]
- ИНГ1 , [ 153 ] [ 154 ]
- ИНГ4 , [ 155 ] [ 156 ]
- ИНГ5 , [ 155 ]
- IκBα , [ 157 ]
- КПНБ1 , [ 139 ]
- ЛМО3 , [ 85 ]
- Мдм2 , [ 120 ] [ 158 ] [ 159 ] [ 160 ]
- МДМ4 , [ 161 ] [ 162 ]
- МЕД1 , [ 163 ] [ 164 ]
- МАПК9 , [ 165 ] [ 166 ]
- МНАТ1 , [ 124 ]
- НДН , [ 167 ]
- НКЛ , [ 168 ]
- ОНЕМЕВШИЙ , [ 169 ]
- NF-κB,[170]
- P16,[126][160][171]
- PARC,[172]
- PARP1,[92][173]
- PIAS1,[128][174]
- CDC14B,[113]
- PIN1,[175][176]
- PLAGL1,[177]
- PLK3,[178][179]
- PRKRA,[180]
- PHB,[181]
- PML,[158][182][183]
- PSME3,[184]
- PTEN,[159]
- PTK2,[185]
- PTTG1,[186]
- RAD51,[102][187][188]
- RCHY1,[189][190]
- RELA,[170]
- Reprimo[citation needed]
- RPA1,[191][192]
- RPL11,[171]
- S100B,[193]
- SUMO1,[194][195]
- SMARCA4,[196]
- SMARCB1,[196]
- SMN1,[197]
- STAT3,[170]
- TBP,[198][199]
- TFAP2A,[200]
- TFDP1,[201]
- TIGAR,[202]
- TOP1,[203][204]
- TOP2A,[205]
- TP53BP1,[103][206][207][208][209][210][211]
- TP53BP2,[211][212]
- TOP2B,[205]
- TP53INP1,[213][214]
- ТСГ101 , [ 215 ]
- БЕ2А , [ 216 ]
- БЭ2И , [ 128 ] [ 194 ] [ 217 ] [ 218 ]
- ЮБК , [ 90 ] [ 184 ] [ 195 ] [ 219 ] [ 220 ] [ 221 ] [ 222 ] [ 223 ]
- УСП7 , [ 224 ]
- USP10 , [ 49 ]
- ВРН , [ 106 ] [ 225 ]
- WWOX , [ 226 ]
- XPB , [ 134 ]
- YBX1 , [ 91 ] [ 227 ]
- ЮПЕЛ3 , [ 228 ]
- ЯВАЗ , [ 229 ]
- Зиф268 , [ 230 ]
- ЗНФ148 , [ 231 ]
- СИРТ1 , [ 232 ]
- циркРНК_014511. [ 233 ]
См. также
[ редактировать ]- Пифитрин , ингибитор Р53.
Примечания
[ редактировать ]Ссылки
[ редактировать ]- ^ Перейти обратно: а б с GRCh38: Версия Ensembl 89: ENSG00000141510 – Ensembl , май 2017 г.
- ^ Перейти обратно: а б с GRCm38: выпуск Ensembl 89: ENSMUSG00000059552 – Ensembl , май 2017 г.
- ^ «Ссылка на Human PubMed:» . Национальный центр биотехнологической информации, Национальная медицинская библиотека США .
- ^ «Ссылка на Mouse PubMed:» . Национальный центр биотехнологической информации, Национальная медицинская библиотека США .
- ^ Перейти обратно: а б с Суржет С., член парламента Хури, Дж. К. Бурдон (декабрь 2013 г.). «Раскрытие роли вариантов сплайсинга р53 в злокачественных новообразованиях человека: клиническая перспектива» . Онкомишени и терапия . 7 : 57–68. дои : 10.2147/OTT.S53876 . ПМЦ 3872270 . ПМИД 24379683 .
- ^ Туфекчан Э, Толедо Ф (май 2018 г.). «Возвращение к «Хранителю генома»: p53 снижает активность генов, необходимых для поддержания теломер, восстановления ДНК и структуры центромер» . Раки . 10 (5): 135. doi : 10.3390/cancers10050135 . ПМЦ 5977108 . ПМИД 29734785 .
- ^ Перейти обратно: а б с Матлашевски Г., Лэмб П., Пим Д. и др. (декабрь 1984 г.). «Выделение и характеристика клона кДНК р53 человека: экспрессия гена р53 человека» . Журнал ЭМБО . 3 (13): 3257–62. дои : 10.1002/j.1460-2075.1984.tb02287.x . ПМЦ 557846 . ПМИД 6396087 .
- ^ Перейти обратно: а б Изобе М., Эмануэль Б.С., Гивол Д. и др. (1986). «Локализация гена опухолевого антигена p53 человека на полосе 17p13». Природа . 320 (6057): 84–5. Бибкод : 1986Natur.320...84I . дои : 10.1038/320084a0 . ПМИД 3456488 . S2CID 4310476 .
- ^ Перейти обратно: а б Керн С.Е., Кинцлер К.В., Брускин А. и др. (июнь 1991 г.). «Идентификация p53 как ДНК-связывающего белка, специфичного для последовательности». Наука . 252 (5013): 1708–11. Бибкод : 1991Sci...252.1708K . дои : 10.1126/science.2047879 . ПМИД 2047879 . S2CID 19647885 .
- ^ Перейти обратно: а б Макбрайд О.В., Мерри Д., Гивол Д. (январь 1986 г.). «Ген клеточного опухолевого антигена p53 человека расположен на коротком плече хромосомы 17 (17p13)» . Труды Национальной академии наук Соединенных Штатов Америки . 83 (1): 130–4. Бибкод : 1986ПНАС...83..130М . дои : 10.1073/pnas.83.1.130 . ПМК 322805 . ПМИД 3001719 .
- ^ Перейти обратно: а б с д Бурдон Дж.К., Фернандес К., Мюррей-Змиевски Ф. и др. (сентябрь 2005 г.). «Изоформы р53 могут регулировать транскрипционную активность р53» . Гены и развитие . 19 (18): 2122–37. дои : 10.1101/gad.1339905 . ПМЦ 1221884 . ПМИД 16131611 .
- ^ Левин А.Дж., Лейн Д.П., ред. (2010). Семейство р53 . Перспективы Колд-Спринг-Харбор в биологии. Колд-Спринг-Харбор, Нью-Йорк: Лабораторное издательство Колд-Спринг-Харбор. ISBN 978-0-87969-830-0 .
- ^ Член парламента Хури, Бурдон Дж.К. Изоформы p53: внутриклеточный микропроцессор? Гены рака. 2011, апрель 2(4): 453-65. дои: 10.1177/1947601911408893. PMID: 21779513; PMCID: PMC3135639.
- ^ Мэй П, Мэй Е (декабрь 1999 г.). «Двадцать лет исследований р53: структурные и функциональные аспекты белка р53» . Онкоген . 18 (53): 7621–36. дои : 10.1038/sj.onc.1203285 . ПМИД 10618702 .
- ^ «Филогенетический маркер OrthoMaM: кодирующая последовательность TP53» . Архивировано из оригинала 17 марта 2018 г. Проверено 2 декабря 2009 г.
- ^ Клуг С.Дж., Рессинг М., Кениг Дж. и др. (август 2009 г.). «Полиморфизм кодона 72 TP53 и рак шейки матки: объединенный анализ отдельных данных из 49 исследований». «Ланцет». Онкология . 10 (8): 772–84. дои : 10.1016/S1470-2045(09)70187-1 . ПМИД 19625214 .
- ^ Сонояма Т., Сакаи А., Мита Ю. и др. (2011). «Полиморфизм кодона 72 TP53 связан с риском рака поджелудочной железы у мужчин, курильщиков и пьющих» . Отчеты о молекулярной медицине . 4 (3): 489–95. дои : 10.3892/ммр.2011.449 . ПМИД 21468597 .
- ^ Алавади С., Габро Л., Алсалех М. и др. (сентябрь 2011 г.). «Полиморфизм гена P53 и риск рака молочной железы у арабских женщин». Медицинская онкология . 28 (3): 709–15. дои : 10.1007/s12032-010-9505-4 . ПМИД 20443084 . S2CID 207372095 .
- ^ Ю Х., Хуан Ю.Дж., Лю Цз. и др. (сентябрь 2011 г.). «Влияние полиморфизма промотора MDM2 и полиморфизма кодона 72 p53 на риск и возраст начала плоскоклеточного рака головы и шеи» . Молекулярный канцерогенез . 50 (9): 697–706. дои : 10.1002/mc.20806 . ПМЦ 3142329 . ПМИД 21656578 .
- ^ Пяо Дж.М., Ким Х.Н., Сонг Х.Р. и др. (сентябрь 2011 г.). «Полиморфизм кодона 72 p53 и риск рака легких у корейского населения». Рак легких . 73 (3): 264–7. дои : 10.1016/j.lungcan.2010.12.017 . ПМИД 21316118 .
- ^ Ван Дж.Дж., Чжэн Ю., Сунь Л. и др. (ноябрь 2011 г.). «Полиморфизм кодона 72 TP53 и предрасположенность к колоректальному раку: метаанализ». Отчеты по молекулярной биологии . 38 (8): 4847–53. дои : 10.1007/s11033-010-0619-8 . ПМИД 21140221 . S2CID 11730631 .
- ^ Цзян Д.К., Яо Л., Рен В.Х. и др. (декабрь 2011 г.). «Полиморфизм TP53 Arg72Pro и риск рака эндометрия: метаанализ». Медицинская онкология . 28 (4): 1129–35. дои : 10.1007/s12032-010-9597-x . ПМИД 20552298 . S2CID 32990396 .
- ^ Туроу Х.С., Хаак Р., Хартвиг Ф.П. и др. (декабрь 2011 г.). «Полиморфизм гена TP53: значение для рака, этнической принадлежности и веса при рождении в бразильской когорте». Журнал биологических наук . 36 (5): 823–31. дои : 10.1007/s12038-011-9147-5 . ПМИД 22116280 . S2CID 23027087 .
- ^ Хуан С.И., Су К.Т., Чу Дж.С. и др. (декабрь 2011 г.). «Полиморфизмы кодона 72 P53 и SNP309 MDM2 и риск почечно-клеточного рака в зоне с низким уровнем воздействия мышьяка». Токсикология и прикладная фармакология . 257 (3): 349–55. дои : 10.1016/j.taap.2011.09.018 . ПМИД 21982800 .
- ^ Гилберт С.Ф. Биология развития, 10-е изд . Сандерленд, Массачусетс, США: Издательство Sinauer Associates, Inc. п. 588.
- ^ Национальный центр биотехнологической информации (1998). «Кожа и соединительная ткань» . Гены и болезни . Национальные институты здравоохранения США . Проверено 28 мая 2008 г.
- ^ Бейтс С., Филлипс А.С., Кларк П.А. и др. (сентябрь 1998 г.). «p14ARF связывает опухолевые супрессоры RB и p53». Природа . 395 (6698): 124–5. Бибкод : 1998Natur.395..124B . дои : 10.1038/25867 . ПМИД 9744267 . S2CID 4355786 .
- ^ «Хранитель Генома начал загорать» . Новый учёный . 17 марта 2007 года . Проверено 29 марта 2007 г.
- ^ Куи Р., Видлунд Х.Р., Файги Э. и др. (март 2007 г.). «Центральная роль р53 в реакции на загар и патологической гиперпигментации» . Клетка . 128 (5): 853–64. дои : 10.1016/j.cell.2006.12.045 . ПМИД 17350573 .
- ^ Перейти обратно: а б Джайн А.К., Олтон К., Яковино М. и др. (2012). «p53 регулирует клеточный цикл и микроРНК, способствуя дифференцировке эмбриональных стволовых клеток человека» . ПЛОС Биология . 10 (2): e1001268. дои : 10.1371/journal.pbio.1001268 . ПМК 3289600 . ПМИД 22389628 .
- ^ Майметс Т., Неганова И., Армстронг Л. и др. (сентябрь 2008 г.). «Активация р53 нутлином приводит к быстрой дифференцировке эмбриональных стволовых клеток человека» . Онкоген . 27 (40): 5277–87. дои : 10.1038/onc.2008.166 . ПМИД 18521083 .
- ^ тер Хуурне М., Пэн Т., Йи Г. и др. (февраль 2020 г.). «Критическая роль P53 в регуляции клеточного цикла эмбриональных стволовых клеток основного состояния» . Отчеты о стволовых клетках . 14 (2): 175–183. doi : 10.1016/j.stemcr.2020.01.001 . ПМК 7013234 . ПМИД 32004494 .
- ^ Дас Б., Баят-Мохтари Р., Цуй М. и др. (август 2012 г.). «HIF-2α подавляет р53, повышая стволовость и регенеративный потенциал эмбриональных стволовых клеток человека» . Стволовые клетки . 30 (8): 1685–95. дои : 10.1002/stem.1142 . ПМЦ 3584519 . ПМИД 22689594 .
- ^ Лейк Б.Б., Финк Дж., Клеметсауне Л. и др. (май 2012 г.). «Контекст-зависимое усиление индуцированного перепрограммирования плюрипотентных стволовых клеток путем подавления Puma» . Стволовые клетки . 30 (5): 888–97. дои : 10.1002/stem.1054 . ПМК 3531606 . ПМИД 22311782 .
- ^ Марион Р.М., Страти К., Ли Х. и др. (август 2009 г.). «Реакция на повреждение ДНК, опосредованная р53, ограничивает перепрограммирование для обеспечения геномной целостности iPS-клеток» . Природа . 460 (7259): 1149–53. Бибкод : 2009Natur.460.1149M . дои : 10.1038/nature08287 . ПМК 3624089 . ПМИД 19668189 .
- ^ Юн М.Х., Гейтс П.Б., Брокс Дж.П. (октябрь 2013 г.). «Регуляция р53 имеет решающее значение для регенерации конечностей позвоночных» . Труды Национальной академии наук Соединенных Штатов Америки . 110 (43): 17392–7. Бибкод : 2013PNAS..11017392Y . дои : 10.1073/pnas.1310519110 . ПМЦ 3808590 . ПМИД 24101460 .
- ^ Алони-Гринштейн Р., Шетцер Ю., Кауфман Т. и др. (август 2014 г.). «p53: барьер на пути образования раковых стволовых клеток» . Письма ФЭБС . 588 (16): 2580–9. дои : 10.1016/j.febslet.2014.02.011 . ПМИД 24560790 . S2CID 37901173 .
- ^ Перейти обратно: а б Бабаи Г., Алиараб А., Асгари Востоколаи М. и др. (ноябрь 2021 г.). «Перекрестная связь между p53 и метастазами: фокус на эпителиально-мезенхимальном переходе, раковых стволовых клетках, ангиогенезе, аутофагии и аноикисе». Отчеты по молекулярной биологии . 48 (11): 7545–7557. дои : 10.1007/s11033-021-06706-1 . ПМИД 34519942 . S2CID 237506513 .
- ^ Теодоро Дж.Г., Эванс С.К., Грин М.Р. (ноябрь 2007 г.). «Ингибирование опухолевого ангиогенеза с помощью р53: новая роль хранителя генома». Журнал молекулярной медицины (обзор). 85 (11): 1175–1186. дои : 10.1007/s00109-007-0221-2 . ПМИД 17589818 . S2CID 10094554 .
- ^ Асадиан С., Эль-Ассаад В., Ван XQ и др. (март 2012 г.). «p53 ингибирует ангиогенез, индуцируя выработку аррестена» . Исследования рака . 72 (5): 1270–1279. дои : 10.1158/0008-5472.CAN-11-2348 . ПМИД 22253229 .
- ^ Ху В., Фэн З., Терески А.К. и др. (ноябрь 2007 г.). «p53 регулирует материнское воспроизводство посредством LIF». Природа . 450 (7170): 721–4. Бибкод : 2007Natur.450..721H . дои : 10.1038/nature05993 . ПМИД 18046411 . S2CID 4357527 .
- ^ Борхсениус С.Н., Дакс А., Федорова О. и др. (январь 2018 г.). «Влияние микоплазменной инфекции на реакцию организма хозяина посредством передачи сигналов p53/NF-κB». Журнал клеточной физиологии . 234 (1): 171–180. дои : 10.1002/jcp.26781 . ПМИД 30146800 .
- ^ Быков В.Я., Эрикссон С.Е., Бьянки Дж. и др. (февраль 2018 г.). «Нацеливание на мутантный р53 для эффективной терапии рака». Обзоры природы. Рак . 18 (2): 89–102. дои : 10.1038/nrc.2017.109 . ПМИД 29242642 . S2CID 4552678 .
- ^ Хан Э.С., Мюллер Ф.Л., Перес VI и др. (июнь 2008 г.). «Признак экспрессии генов in vivo, вызывающий окислительный стресс» . Физиологическая геномика . 34 (1): 112–126. doi : 10.1152/физиологгеномика.00239.2007 . ПМЦ 2532791 . ПМИД 18445702 .
- ^ Грахалес-Рейес Г.Е., Колонна М. (август 2020 г.). «Интерфероновые реакции при вирусных пневмониях». Наука . 369 (6504): 626–627. Бибкод : 2020Sci...369..626G . дои : 10.1126/science.abd2208 . ПМИД 32764056 .
- ^ Первис Дж.Э., Кархос К.В., Мок С. и др. (июнь 2012 г.). «Динамика p53 контролирует судьбу клеток» . Наука . 336 (6087): 1440–1444. Бибкод : 2012Sci...336.1440P . дои : 10.1126/science.1218351 . ПМК 4162876 . ПМИД 22700930 .
- ^ Каннер Дж.А., Собо М., Болл С. и др. (сентябрь 2009 г.). «МИ-63: новый низкомолекулярный ингибитор нацелен на MDM2 и индуцирует апоптоз в эмбриональных и альвеолярных клетках рабдомиосаркомы с р53 дикого типа» . Британский журнал рака . 101 (5): 774–81. дои : 10.1038/sj.bjc.6605199 . ПМЦ 2736841 . ПМИД 19707204 .
- ^ Хок А.К., Виньерон А.М., Картер С. и др. (ноябрь 2011 г.). «Регуляция стабильности и функции р53 с помощью деубиквитинирующего фермента USP42» . Журнал ЭМБО . 30 (24): 4921–30. дои : 10.1038/emboj.2011.419 . ПМЦ 3243628 . ПМИД 22085928 .
- ^ Перейти обратно: а б Юань Дж., Луо К., Чжан Л. и др. (февраль 2010 г.). «USP10 регулирует локализацию и стабильность p53 путем деубиквитинирования p53» . Клетка . 140 (3): 384–396. дои : 10.1016/j.cell.2009.12.032 . ПМК 2820153 . ПМИД 20096447 .
- ^ Вахрушева О., Смолка С., Гаджавада П. и др. (март 2008 г.). «Sirt7 повышает стрессоустойчивость кардиомиоцитов и предотвращает апоптоз и воспалительную кардиомиопатию у мышей» . Исследование кровообращения . 102 (6): 703–10. дои : 10.1161/CIRCRESAHA.107.164558 . ПМИД 18239138 .
- ^ Исбель Л., Искар М., Дурду С. и др. (июнь 2023 г.). «Считывание метилирования гистонов с помощью Trim24 локально ограничивает открытие хроматина с помощью p53» . Структурная и молекулярная биология природы . 30 (7): 948–57. дои : 10.1038/s41594-023-01021-8 . ПМИД 37386214 .
- ^ Холлштейн М., Сидранский Д., Фогельштейн Б. и др. (июль 1991 г.). «Мутации р53 при раке человека» . Наука . 253 (5015): 49–53. Бибкод : 1991Sci...253...49H . дои : 10.1126/science.1905840 . ПМИД 1905840 . S2CID 38527914 .
- ^ Шмитт К.А., Фридман Дж.С., Ян М. и др. (апрель 2002 г.). «Раскрытие функций супрессора опухоли p53 in vivo» . Раковая клетка . 1 (3): 289–98. дои : 10.1016/S1535-6108(02)00047-8 . ПМИД 12086865 .
- ^ Тайнер С.Д., Венкатачалам С., Чой Дж. и др. (январь 2002 г.). «Мутантные мыши p53, демонстрирующие фенотипы, связанные с ранним старением». Природа . 415 (6867): 45–53. Бибкод : 2002Natur.415...45T . дои : 10.1038/415045а . ПМИД 11780111 . S2CID 749047 .
- ^ Вентура А., Кирш Д.Г., Маклафлин М.Е. и др. (февраль 2007 г.). «Восстановление функции р53 приводит к регрессии опухоли in vivo». Природа . 445 (7128): 661–5. дои : 10.1038/nature05541 . ПМИД 17251932 . S2CID 4373520 .
- ^ Герце Х.Д., Денг В., Хельма Дж. и др. (2013). «Визуализация и целенаправленное нарушение белковых взаимодействий в живых клетках» . Природные коммуникации . 4 : 2660. Бибкод : 2013NatCo...4.2660H . дои : 10.1038/ncomms3660 . ПМЦ 3826628 . ПМИД 24154492 .
- ^ Пирсон С., Цзя Х., Кандачи К. (январь 2004 г.). «Китай одобряет первую генную терапию» . Природная биотехнология . 22 (1): 3–4. дои : 10.1038/nbt0104-3 . ПМК 7097065 . ПМИД 14704685 .
- ^ Анджелетти ПК, Чжан Л., Вуд С. (2008). «Вирусная этиология злокачественных опухолей, связанных со СПИДом». ВИЧ-1: молекулярная биология и патогенез . Достижения фармакологии. Том. 56. стр. 509–57. дои : 10.1016/S1054-3589(07)56016-3 . ISBN 978-0-12-373601-7 . ПМК 2149907 . ПМИД 18086422 .
- ^ Рибейро А.С., Шарлебуа Д.А., Ллойд-Прайс Дж. (декабрь 2007 г.). «CellLine, симулятор стохастического клеточного происхождения» . Биоинформатика . 23 (24): 3409–3411. doi : 10.1093/биоинформатика/btm491 . ПМИД 17928303 .
- ^ Перейти обратно: а б Буллок А.Н., Хенкель Дж., ДеДекер Б.С. и др. (декабрь 1997 г.). «Термодинамическая стабильность корового домена р53 дикого типа и мутанта» . Труды Национальной академии наук Соединенных Штатов Америки . 94 (26): 14338–42. Бибкод : 1997PNAS...9414338B . дои : 10.1073/pnas.94.26.14338 . ПМК 24967 . ПМИД 9405613 .
- ^ Кебель М., Роннетт Б.М., Сингх Н. и др. (январь 2019 г.). «Интерпретация иммуногистохимии P53 при карциноме эндометрия: на пути к повышению воспроизводимости» . Международный журнал гинекологической патологии . 38 (Приложение 1): S123–S131. дои : 10.1097/PGP.0000000000000488 . ПМК 6127005 . ПМИД 29517499 .
 В эту статью включен текст, доступный по лицензии CC BY 4.0 .
В эту статью включен текст, доступный по лицензии CC BY 4.0 .
- ^ Изображение взято из следующего источника с некоторыми изменениями Микаэля Хэггстрёма, доктора медицинских наук:
- Шалленберг С., Плаж Х., Хофбауэр С. и др. (2023). «Измененная экспрессия p53/p16 связана с прогрессированием уротелиальной карциномы, но в значительной степени не связана с прогнозом при мышечно-инвазивных опухолях» . Акта Онкол . 62 (12): 1880–1889. дои : 10.1080/0284186X.2023.2277344 . ПМИД 37938166 . - ^ Источник роли в различении PUNLMP от рака низкой степени злокачественности:
- Калантари М.Р., Ахмадния Х (2007). «Сверхэкспрессия P53 при уротелиальных новообразованиях мочевого пузыря: новый аспект классификации Всемирной организации здравоохранения / Международного общества урологической патологии» . Урол Дж . 4 (4): 230–3. ПМИД 18270948 . - ^ Миткин Н.А., Хук К.Д., Шварц А.М. и др. (март 2015 г.). «p53-зависимая экспрессия хемокинового рецептора CXCR5 в клетках рака молочной железы MCF-7» . Научные отчеты . 5 (5): 9330. Бибкод : 2015NatSR...5E9330M . дои : 10.1038/srep09330 . ПМК 4365401 . ПМИД 25786345 .
- ^ Авраам С.А., Хопкрофт Л.Е., Каррик Э. и др. (июнь 2016 г.). «Двойное воздействие на p53 и c-MYC избирательно уничтожает лейкозные стволовые клетки» . Природа . 534 (7607): 341–6. Бибкод : 2016Natur.534..341A . дои : 10.1038/nature18288 . ПМЦ 4913876 . ПМИД 27281222 .
- ^ «Ученые идентифицируют лекарства, воздействующие на «ахиллесову пяту» клеток хронического миелолейкоза» . мояНаука . 08.06.2016 . Проверено 9 июня 2016 г.
- ^ Перейти обратно: а б Член парламента от Хури, Дж. К. Бурдон (апрель 2011 г.). «Изоформы p53: внутриклеточный микропроцессор?» . Гены и рак . 2 (4): 453–65. дои : 10.1177/1947601911408893 . ПМЦ 3135639 . ПМИД 21779513 .
- ^ Эйвери-Кейда К.А., Мортен Б., Вонг-Браун М.В. и др. (март 2014 г.). «Относительная экспрессия мРНК изоформ р53 при раке молочной железы связана с клиническими особенностями и исходом» . Канцерогенез . 35 (3): 586–96. дои : 10.1093/carcin/bgt411 . ПМИД 24336193 .
- ^ Арсик Н., Гадеа Г., Лагерквист Э.Л. и др. (апрель 2015 г.). «Изоформа p53 Δ133p53β повышает потенциал раковых стволовых клеток» . Отчеты о стволовых клетках . 4 (4): 531–40. дои : 10.1016/j.stemcr.2015.02.001 . ПМК 4400643 . ПМИД 25754205 .
- ^ Харами-Папп Х., Понгор Л.С., Мункачи Г. и др. (октябрь 2016 г.). «Мутация TP53 влияет на энергетический обмен и увеличивает гликолиз при раке молочной железы» . Онкотаргет . 7 (41): 67183–67195. дои : 10.18632/oncotarget.11594 . ПМК 5341867 . ПМИД 27582538 .
- ^ Гева-Заторский Н., Розенфельд Н., Ицковиц С. и др. (июнь 2006 г.). «Колебания и изменчивость в системе р53» . Молекулярная системная биология . 2 : 2006.0033. дои : 10.1038/msb4100068 . ПМК 1681500 . ПМИД 16773083 .
- ^ Проктор CJ, Грей DA (август 2008 г.). «Объяснение колебаний и изменчивости в системе p53-Mdm2» . Системная биология BMC . 2 (75): 75. дои : 10.1186/1752-0509-2-75 . ПМЦ 2553322 . ПМИД 18706112 .
- ^ Чонг К.Х., Самарасингхе С., Куласири Д. (декабрь 2013 г.). «Математическое моделирование базальной динамики р53 и реакции на повреждение ДНК». С-фаКС . 259 (20-й Международный конгресс по математическому моделированию и симуляции): 670–6. дои : 10.1016/j.mbs.2014.10.010 . ПМИД 25433195 .
- ^ Чумаков П.М., Иотсова В.С., Георгиев Г.П. (1982). «[Выделение плазмидного клона, содержащего последовательность мРНК мышиного невирусного Т-антигена]». Доклады Академии наук СССР . 267 (5): 1272–5. ПМИД 6295732 .
- ^ Орен М., Левин А.Дж. (январь 1983 г.). «Молекулярное клонирование кДНК, специфичной для мышиного клеточного опухолевого антигена p53» . Труды Национальной академии наук Соединенных Штатов Америки . 80 (1): 56–9. Бибкод : 1983PNAS...80...56O . дои : 10.1073/pnas.80.1.56 . ПМК 393308 . ПМИД 6296874 .
- ^ Закут-Хури Р., Орен М., Биенц Б. и др. (1983). «Один ген и псевдоген клеточного опухолевого антигена p53». Природа . 306 (5943): 594–7. Бибкод : 1983Natur.306..594Z . дои : 10.1038/306594a0 . ПМИД 6646235 . S2CID 4325094 .
- ^ Закут-Хури Р., Биенц-Тадмор Б., Гивол Д. и др. (май 1985 г.). «Человеческий клеточный опухолевый антиген p53: последовательность и экспрессия кДНК в клетках COS» . Журнал ЭМБО . 4 (5): 1251–5. дои : 10.1002/j.1460-2075.1985.tb03768.x . ПМЦ 554332 . ПМИД 4006916 .
- ^ Бейкер С.Дж., Фирон Э.Р., Нигро Дж.М. и др. (апрель 1989 г.). «Делеции 17 хромосомы и мутации гена p53 при колоректальном раке». Наука . 244 (4901): 217–21. Бибкод : 1989Sci...244..217B . дои : 10.1126/science.2649981 . ПМИД 2649981 .
- ^ Финлей Калифорния, Хиндс П.В., Левин А.Дж. (июнь 1989 г.). «Протоонкоген р53 может действовать как супрессор трансформации» . Клетка . 57 (7): 1083–93. дои : 10.1016/0092-8674(89)90045-7 . ПМИД 2525423 .
- ^ Рэйкрофт Л., Ву ХИ, Лозано Дж. (август 1990 г.). «Активация транскрипции диким типом, но не трансформирующими мутантами антионкогена p53» . Наука . 249 (4972): 1049–1051. Бибкод : 1990Sci...249.1049R . дои : 10.1126/science.2144364 . ПМЦ 2935288 . ПМИД 2144364 .
- ^ Мальцман В., Чижик Л. (сентябрь 1984 г.). «УФ-облучение стимулирует уровни клеточного опухолевого антигена p53 в нетрансформированных клетках мыши» . Молекулярная и клеточная биология . 4 (9): 1689–94. дои : 10.1128/mcb.4.9.1689 . ПМЦ 368974 . ПМИД 6092932 .
- ^ Кастан М.Б., Куэрбитц С.Дж. (декабрь 1993 г.). «Контроль ареста G1 после повреждения ДНК» . Перспективы гигиены окружающей среды . 101 (Приложение 5): 55–8. дои : 10.2307/3431842 . JSTOR 3431842 . ПМЦ 1519427 . ПМИД 8013425 .
- ^ Кошланд Д.Э. (декабрь 1993 г.). «Молекула года». Наука . 262 (5142): 1953. Бибкод : 1993Sci...262.1953K . дои : 10.1126/science.8266084 . ПМИД 8266084 .
- ^ Вено С., Маратрат М., Дюрей С. и др. (август 1998 г.). «Потребность в функциональном домене p53, богатом пролином, для опосредования апоптоза коррелирует со специфической трансактивацией гена PIG3 и с репрессией транскрипции» . Журнал ЭМБО . 17 (16): 4668–79. дои : 10.1093/emboj/17.16.4668 . ПМЦ 1170796 . ПМИД 9707426 .
- ^ Перейти обратно: а б Ларсен С., Йокочи Т., Исогай Э. и др. (февраль 2010 г.). «LMO3 взаимодействует с p53 и ингибирует его транскрипционную активность». Связь с биохимическими и биофизическими исследованиями . 392 (3): 252–7. дои : 10.1016/j.bbrc.2009.12.010 . ПМИД 19995558 .
- ^ Хармс К.Л., Чен X (март 2005 г.). «С-конец белков семейства р53 является определяющим фактором судьбы клетки» . Молекулярная и клеточная биология . 25 (5): 2014–30. дои : 10.1128/MCB.25.5.2014-2030.2005 . ПМК 549381 . ПМИД 15713654 .
- ^ Белл С., Кляйн С., Мюллер Л. и др. (октябрь 2002 г.). «p53 содержит большие неструктурированные области в исходном состоянии». Журнал молекулярной биологии . 322 (5): 917–27. дои : 10.1016/S0022-2836(02)00848-3 . ПМИД 12367518 .
- ^ Цимер М.А., Мейсон А., Карлсон Д.М. (сентябрь 1982 г.). «Бесклеточные трансляции мРНК белков, богатых пролином» . Журнал биологической химии . 257 (18): 11176–80. дои : 10.1016/S0021-9258(18)33948-6 . ПМИД 7107651 .
- ^ Чжу Дж., Чжан С., Цзян Дж. и др. (декабрь 2000 г.). «Определение функциональных доменов р53, необходимых для индукции апоптоза» . Журнал биологической химии . 275 (51): 39927–34. дои : 10.1074/jbc.M005676200 . ПМИД 10982799 .
- ^ Перейти обратно: а б Хан Дж.М., Пак Б.Дж., Пак С.Г. и др. (август 2008 г.). «AIMP2/p38, каркас для мульти-тРНК-синтетазного комплекса, реагирует на генотоксический стресс через p53» . Труды Национальной академии наук Соединенных Штатов Америки . 105 (32): 11206–11. Бибкод : 2008PNAS..10511206H . дои : 10.1073/pnas.0800297105 . ПМК 2516205 . ПМИД 18695251 .
- ^ Перейти обратно: а б Койич С., Медеот Е., Гуччионе Е. и др. (май 2004 г.). «Белок Ankrd2, связь между саркомером и ядром скелетных мышц». Журнал молекулярной биологии . 339 (2): 313–25. дои : 10.1016/j.jmb.2004.03.071 . ПМИД 15136035 .
- ^ Перейти обратно: а б Гувен Н., Бешерель О.Дж., Киджас А.В. и др. (май 2004 г.). «Апратаксин, новый белок, защищающий от генотоксического стресса» . Молекулярная генетика человека . 13 (10): 1081–93. дои : 10.1093/hmg/ddh122 . ПМИД 15044383 .
- ^ Перейти обратно: а б Фаббро М., Сэвидж К., Хобсон К. и др. (июль 2004 г.). «Комплексы BRCA1-BARD1 необходимы для фосфорилирования p53Ser-15 и остановки G1/S после повреждения ДНК, вызванного ионизирующим излучением» . Журнал биологической химии . 279 (30): 31251–8. дои : 10.1074/jbc.M405372200 . ПМИД 15159397 .
- ^ Перейти обратно: а б с Ким С.Т., Лим Д.С., Канман С.Э. и др. (декабрь 1999 г.). «Специфичность субстратов и идентификация предполагаемых субстратов членов семейства ATM-киназ» . Журнал биологической химии . 274 (53): 37538–43. дои : 10.1074/jbc.274.53.37538 . ПМИД 10608806 .
- ^ Кан Дж., Фергюсон Д., Сонг Х. и др. (январь 2005 г.). «Функциональное взаимодействие H2AX, NBS1 и p53 в АТМ-зависимых реакциях на повреждение ДНК и подавлении опухоли» . Молекулярная и клеточная биология . 25 (2): 661–70. дои : 10.1128/MCB.25.2.661-670.2005 . ПМК 543410 . ПМИД 15632067 .
- ^ Ханна К.К., Китинг К.Е., Козлов С. и др. (декабрь 1998 г.). «ATM связывается с p53 и фосфорилирует: картирование области взаимодействия». Природная генетика . 20 (4): 398–400. дои : 10.1038/3882 . ПМИД 9843217 . S2CID 23994762 .
- ^ Вестфаль С.Х., Шмальц С., Роуэн С. и др. (май 1997 г.). «Генетические взаимодействия между atm и p53 влияют на клеточную пролиферацию и контрольные точки клеточного цикла, индуцированные облучением». Исследования рака . 57 (9): 1664–7. ПМИД 9135004 .
- ^ Стельзл Ю., Ворм Ю., Лаловски М. и др. (сентябрь 2005 г.). «Сеть белок-белкового взаимодействия человека: ресурс для аннотирования протеома» . Клетка . 122 (6): 957–68. дои : 10.1016/j.cell.2005.08.029 . hdl : 11858/00-001M-0000-0010-8592-0 . ПМИД 16169070 .
- ^ Ян С., Ван Х., Бойд Д.Д. (март 2002 г.). «ATF3 подавляет экспрессию коллагеназы IV типа (MMP-2) массой 72 кДа, противодействуя p53-зависимой трансактивации промотора коллагеназы» . Журнал биологической химии . 277 (13): 10804–12. дои : 10.1074/jbc.M112069200 . ПМИД 11792711 .
- ^ Чен С.С., Чанг ПК, Ченг Ю.В. и др. (сентябрь 2002 г.). «Подавление онкогенной активности STK15 требует независимой от трансактивации функции р53» . Журнал ЭМБО . 21 (17): 4491–9. дои : 10.1093/emboj/cdf409 . ПМК 126178 . ПМИД 12198151 .
- ^ Леу Дж.И., Дюмон П., Хафи М. и др. (май 2004 г.). «Митохондриальный p53 активирует Bak и вызывает разрушение комплекса Bak-Mcl1». Природная клеточная биология . 6 (5): 443–50. дои : 10.1038/ncb1123 . ПМИД 15077116 . S2CID 43063712 .
- ^ Перейти обратно: а б с д и ж Донг Ю, Хакими М.А., Чен X и др. (ноябрь 2003 г.). «Регуляция BRCC, голоферментного комплекса, содержащего BRCA1 и BRCA2, с помощью сигналосомоподобной субъединицы и ее роль в репарации ДНК» . Молекулярная клетка . 12 (5): 1087–99. дои : 10.1016/S1097-2765(03)00424-6 . ПМИД 14636569 .
- ^ Перейти обратно: а б с Сенгупта С., Роблес А.И., Линке С.П. и др. (сентябрь 2004 г.). «Функциональное взаимодействие между хеликазой BLM и 53BP1 в Chk1-опосредованном пути во время ареста S-фазы» . Журнал клеточной биологии . 166 (6): 801–13. дои : 10.1083/jcb.200405128 . ПМК 2172115 . ПМИД 15364958 .
- ^ Ван XW, Ценг А., Эллис Н.А. и др. (август 2001 г.). «Функциональное взаимодействие р53 и ДНК-хеликазы BLM при апоптозе» . Журнал биологической химии . 276 (35): 32948–55. дои : 10.1074/jbc.M103298200 . ПМИД 11399766 .
- ^ Гаркавцев ИВ, Клей Н, Григорян ИА и др. (декабрь 2001 г.). «Белок синдрома Блума взаимодействует и сотрудничает с р53 в регуляции транскрипции и контроле роста клеток». Онкоген . 20 (57): 8276–80. дои : 10.1038/sj.onc.1205120 . ПМИД 11781842 . S2CID 13084911 .
- ^ Перейти обратно: а б Ян Ц., Чжан Р., Ван XW и др. (август 2002 г.). «Обработка соединений Холлидея хеликазами BLM и WRN регулируется р53» . Журнал биологической химии . 277 (35): 31980–7. дои : 10.1074/jbc.M204111200 . hdl : 10026.1/10341 . ПМИД 12080066 .
- ^ Абрамович С., Вернер Х. (2003). «Функциональные и физические взаимодействия между BRCA1 и p53 в регуляции транскрипции гена IGF-IR». Гормональные и метаболические исследования . 35 (11–12): 758–62. дои : 10.1055/s-2004-814154 . ПМИД 14710355 . S2CID 20898175 .
- ^ Оучи Т., Монтейро А.Н., Август А. и др. (март 1998 г.). «BRCA1 регулирует p53-зависимую экспрессию генов» . Труды Национальной академии наук Соединенных Штатов Америки . 95 (5): 2302–6. Бибкод : 1998PNAS...95.2302O . дои : 10.1073/pnas.95.5.2302 . ЧВК 19327 . ПМИД 9482880 .
- ^ Чай Ю.Л., Цуй Дж., Шао Н. и др. (январь 1999 г.). «Второй домен BRCT белков BRCA1 взаимодействует с p53 и стимулирует транскрипцию с промотора p21WAF1/CIP1». Онкоген . 18 (1): 263–8. дои : 10.1038/sj.onc.1202323 . ПМИД 9926942 . S2CID 7462625 .
- ^ Чжан Х., Сомасундарам К., Пэн Ю. и др. (апрель 1998 г.). «BRCA1 физически связывается с p53 и стимулирует его транскрипционную активность». Онкоген . 16 (13): 1713–21. дои : 10.1038/sj.onc.1201932 . ПМИД 9582019 . S2CID 24616900 .
- ^ Марморштейн Л.Ю., Оучи Т., Ааронсон С.А. (ноябрь 1998 г.). «Продукт гена BRCA2 функционально взаимодействует с p53 и RAD51» . Труды Национальной академии наук Соединенных Штатов Америки . 95 (23): 13869–74. Бибкод : 1998PNAS...9513869M . дои : 10.1073/pnas.95.23.13869 . ПМК 24938 . ПМИД 9811893 .
- ^ Урамото Х., Идзуми Х., Нагатани Г. и др. (апрель 2003 г.). «Физическое взаимодействие опухолевого супрессора p53/p73 с CCAAT-связывающим транскрипционным фактором 2 (CTF2) и дифференциальная регуляция экспрессии гена группы 1 человека с высокой подвижностью (HMG1)» . Биохимический журнал . 371 (Часть 2): 301–10. дои : 10.1042/BJ20021646 . ПМЦ 1223307 . ПМИД 12534345 .
- ^ Перейти обратно: а б Ли Л., Юнгман М., Диксон Дж.Э. (январь 2000 г.). «Человеческие фосфатазы Cdc14 взаимодействуют с белком-супрессором опухоли p53 и дефосфорилируют его» . Журнал биологической химии . 275 (4): 2410–4. дои : 10.1074/jbc.275.4.2410 . ПМИД 10644693 .
- ^ Лучани М.Г., Хатчинс Дж.Р., Желева Д. и др. (июль 2000 г.). «С-концевой регуляторный домен р53 содержит функциональный сайт стыковки циклина А». Журнал молекулярной биологии . 300 (3): 503–18. дои : 10.1006/jmbi.2000.3830 . ПМИД 10884347 .
- ^ Абабне М., Гетц С., Монтенарх М. (май 2001 г.). «Понижение активности протеинкиназы cdc2/циклин B путем связывания p53 с p34 (cdc2)». Связь с биохимическими и биофизическими исследованиями . 283 (2): 507–12. дои : 10.1006/bbrc.2001.4792 . ПМИД 11327730 .
- ^ Абедини М.Р., Мюллер Э.Дж., Брун Дж. и др. (июнь 2008 г.). «Цисплатин индуцирует p53-зависимое убиквитинирование FLICE-подобного ингибирующего белка в клетках рака яичников» . Исследования рака . 68 (12): 4511–7. дои : 10.1158/0008-5472.CAN-08-0673 . ПМИД 18559494 .
- ^ Перейти обратно: а б Гудлок Д.М., Цзян К., Перейра Э. и др. (август 2003 г.). «Регуляторные взаимодействия между киназой контрольной точки Chk1 и белками ДНК-зависимого протеинкиназного комплекса» . Журнал биологической химии . 278 (32): 29940–7. дои : 10.1074/jbc.M301765200 . ПМИД 12756247 .
- ^ Тиан Х., Фадже А.Т., Ли С.Л. и др. (2002). «Радиационно-индуцированное фосфорилирование Chk1 по S345 связано с p53-зависимыми путями остановки клеточного цикла» . Неоплазия . 4 (2): 171–80. дои : 10.1038/sj.neo.7900219 . ПМК 1550321 . ПМИД 11896572 .
- ^ Чжао Л., Сэмюэлс Т., Винклер С. и др. (январь 2003 г.). «Циклин G1 обладает ингибирующей рост активностью, связанной с путями супрессора опухолей ARF-Mdm2-p53 и pRb». Молекулярные исследования рака . 1 (3): 195–206. ПМИД 12556559 .
- ^ Перейти обратно: а б Ито А., Кавагути Ю., Лай Ч. и др. (ноябрь 2002 г.). «Для его деградации необходимо деацетилирование р53, опосредованное MDM2-HDAC1» . Журнал ЭМБО . 21 (22): 6236–45. дои : 10.1093/emboj/cdf616 . ПМК 137207 . ПМИД 12426395 .
- ^ Перейти обратно: а б Ливенгуд Дж.А., Скоггин К.Е., Ван Орден К. и др. (март 2002 г.). «Транскрипционная активность p53 опосредуется через взаимодействующий с SRC1 домен CBP/p300» . Журнал биологической химии . 277 (11): 9054–61. дои : 10.1074/jbc.M108870200 . ПМИД 11782467 .
- ^ Перейти обратно: а б Гиблер Х.А., Лемассон И., Нюборг Дж.К. (июль 2000 г.). «Привлечение p53 CREB-связывающего белка, опосредованное фосфорилированным CREB: новый путь регуляции опухолевого супрессора» . Молекулярная и клеточная биология . 20 (13): 4849–58. дои : 10.1128/MCB.20.13.4849-4858.2000 . ПМК 85936 . ПМИД 10848610 .
- ^ Перейти обратно: а б Шнайдер Э., Монтенар М., Вагнер П. (ноябрь 1998 г.). «Регуляция активности киназы CAK с помощью p53». Онкоген . 17 (21): 2733–41. дои : 10.1038/sj.onc.1202504 . ПМИД 9840937 . S2CID 6281777 .
- ^ Перейти обратно: а б Ко Л.Дж., Ши С.Ю., Чен X и др. (декабрь 1997 г.). «p53 фосфорилируется CDK7-циклином H p36MAT1-зависимым образом» . Молекулярная и клеточная биология . 17 (12): 7220–9. дои : 10.1128/mcb.17.12.7220 . ПМК 232579 . ПМИД 9372954 .
- ^ Явузер Ю., Смит Г.К., Блисс Т. и др. (июль 1998 г.). «Независимая от конца ДНК активация ДНК-РК, опосредованная ассоциацией с ДНК-связывающим белком C1D» . Гены и развитие . 12 (14): 2188–99. дои : 10.1101/gad.14.12.2188 . ПМК 317006 . ПМИД 9679063 .
- ^ Перейти обратно: а б Ризос Х., Дифенбах Э., Бадхвар П. и др. (февраль 2003 г.). «Ассоциация p14ARF с репрессором транскрипции p120E4F усиливает ингибирование клеточного цикла» . Журнал биологической химии . 278 (7): 4981–9. дои : 10.1074/jbc.M210978200 . ПМИД 12446718 .
- ^ Сэнди П., Гостисса М., Фогал В. и др. (январь 2000 г.). «p53 участвует в остановке роста, опосредованной p120E4F» . Онкоген . 19 (2): 188–99. дои : 10.1038/sj.onc.1203250 . ПМИД 10644996 .
- ^ Перейти обратно: а б с Галлахер В.М., Аргентини М., Сьерра В. и др. (июнь 1999 г.). «MBP1: новый мутантный белок-партнер, специфичный для p53, с онкогенными свойствами» . Онкоген . 18 (24): 3608–16. дои : 10.1038/sj.onc.1202937 . ПМИД 10380882 .
- ^ Каддихи А.Р., Вонг А.Х., Тэм Н.В. и др. (апрель 1999 г.). «Двухцепочечная РНК-активированная протеинкиназа PKR физически связывается с белком-супрессором опухоли p53 и фосфорилирует p53 человека по серину 392 in vitro». Онкоген . 18 (17): 2690–702. дои : 10.1038/sj.onc.1202620 . ПМИД 10348343 . S2CID 22467088 .
- ^ Синобу Н., Маэда Т., Асо Т. и др. (июнь 1999 г.). «Физическое взаимодействие и функциональный антагонизм между фактором элонгации РНК-полимеразы II и p53» . Журнал биологической химии . 274 (24): 17003–10. дои : 10.1074/jbc.274.24.17003 . ПМИД 10358050 .
- ^ Гроссман С.Р., Перес М., Кунг А.Л. и др. (октябрь 1998 г.). «Комплексы p300/MDM2 участвуют в MDM2-опосредованной деградации p53» . Молекулярная клетка . 2 (4): 405–15. дои : 10.1016/S1097-2765(00)80140-9 . ПМИД 9809062 .
- ^ Ан В., Ким Дж., Редер Р.Г. (июнь 2004 г.). «Упорядоченные совместные функции PRMT1, p300 и CARM1 при активации транскрипции с помощью p53» . Клетка . 117 (6): 735–48. дои : 10.1016/j.cell.2004.05.009 . ПМИД 15186775 .
- ^ Пасторчич М., Дас Х.К. (ноябрь 2000 г.). «Регуляция транскрипции гена пресенилина-1 человека с помощью факторов транскрипции ets и протоонкогена p53» . Журнал биологической химии . 275 (45): 34938–45. дои : 10.1074/jbc.M005411200 . ПМИД 10942770 .
- ^ Перейти обратно: а б Ван XW, Йе Х., Шеффер Л. и др. (июнь 1995 г.). «Модуляция p53 активности эксцизионной репарации нуклеотидов, связанной с TFIIH» . Природная генетика . 10 (2): 188–95. дои : 10.1038/ng0695-188 . hdl : 1765/54884 . ПМИД 7663514 . S2CID 38325851 .
- ^ Ю А, Фань Х.И., Ляо Д. и др. (май 2000 г.). «Активация р53 или потеря белка репарации группы B синдрома Коккейна вызывает метафазную хрупкость человеческих генов U1, U2 и 5S» . Молекулярная клетка . 5 (5): 801–10. дои : 10.1016/S1097-2765(00)80320-2 . ПМИД 10882116 .
- ^ Цай Р.Ю., Маккей Р.Д. (декабрь 2002 г.). «Ядрышковый механизм, контролирующий пролиферацию клеток в стволовых и раковых клетках» . Гены и развитие . 16 (23): 2991–3003. дои : 10.1101/gad.55671 . ЧВК 187487 . ПМИД 12464630 .
- ^ Пэн Ю.К., Куо Ф., Брейдинг Д.Э. и др. (сентябрь 2001 г.). «AMF1 (GPS2) модулирует трансактивацию р53» . Молекулярная и клеточная биология . 21 (17): 5913–24. дои : 10.1128/MCB.21.17.5913-5924.2001 . ПМК 87310 . ПМИД 11486030 .
- ^ Вочарасит П., Бижур Г.Н., Змиевский Дж.В. и др. (июнь 2002 г.). «Прямое, активирующее взаимодействие между киназой гликогенсинтазы-3бета и р53 после повреждения ДНК» . Труды Национальной академии наук Соединенных Штатов Америки . 99 (12): 7951–5. Бибкод : 2002PNAS...99.7951W . дои : 10.1073/pnas.122062299 . ПМК 123001 . ПМИД 12048243 .
- ^ Перейти обратно: а б Акакура С., Ёсида М., Йонеда Й. и др. (май 2001 г.). «Роль Hsc70 в регуляции нуклеоцитоплазматического транспорта чувствительного к температуре p53 (p53Val-135)» . Журнал биологической химии . 276 (18): 14649–57. дои : 10.1074/jbc.M100200200 . ПМИД 11297531 .
- ^ Ван С., Чен Дж. (январь 2003 г.). «Фосфорилирование и связывание hsp90 опосредуют стабилизацию p53 при тепловом шоке» . Журнал биологической химии . 278 (3): 2066–71. дои : 10.1074/jbc.M206697200 . ПМИД 12427754 .
- ^ Пэн Ю, Чен Л, Ли С и др. (ноябрь 2001 г.). «Ингибирование MDM2 с помощью hsp90 способствует стабилизации мутантного р53» . Журнал биологической химии . 276 (44): 40583–90. дои : 10.1074/jbc.M102817200 . ПМИД 11507088 .
- ^ Чен Д., Ли М., Луо Дж. и др. (апрель 2003 г.). «Прямое взаимодействие между HIF-1 альфа и Mdm2 модулирует функцию p53» . Журнал биологической химии . 278 (16): 13595–8. дои : 10.1074/jbc.C200694200 . ПМИД 12606552 .
- ^ Рави Р., Мукерджи Б., Бхуджвалла З.М. и др. (январь 2000 г.). «Регуляция опухолевого ангиогенеза посредством p53-индуцированной деградации индуцируемого гипоксией фактора 1альфа» . Гены и развитие . 14 (1): 34–44. дои : 10.1101/gad.14.1.34 . ПМК 316350 . ПМИД 10640274 .
- ^ Ханссон Л.О., Фридлер А., Фрейнд С. и др. (август 2002 г.). «Два мотива последовательности HIF-1альфа связываются с сайтом связывания ДНК р53» . Труды Национальной академии наук Соединенных Штатов Америки . 99 (16): 10305–9. Бибкод : 2002PNAS...9910305H . дои : 10.1073/pnas.122347199 . ПМК 124909 . ПМИД 12124396 .
- ^ РГ, Канекал М., Саймон М.К. и др. (март 1998 г.). «Стабилизация p53 дикого типа с помощью индуцируемого гипоксией фактора 1альфа». Природа . 392 (6674): 405–8. Бибкод : 1998Natur.392..405A . дои : 10.1038/32925 . ПМИД 9537326 . S2CID 4423081 .
- ^ Кондо С., Лу Ю., Деббас М. и др. (апрель 2003 г.). «Характеристика клеток и мышей, нацеленных на гены, с дефицитом p53-связывающей киназы, взаимодействующей с гомеодоменом протеинкиназы 1 (HIPK1)» . Труды Национальной академии наук Соединенных Штатов Америки . 100 (9): 5431–6. Бибкод : 2003PNAS..100.5431K . дои : 10.1073/pnas.0530308100 . ПМК 154362 . ПМИД 12702766 .
- ^ Хофманн Т.Г., Мёллер А., Сирма Х. и др. (январь 2002 г.). «Регуляция активности р53 путем его взаимодействия с протеинкиназой-2, взаимодействующей с гомеодоменом». Природная клеточная биология . 4 (1): 1–10. дои : 10.1038/ncb715 . ПМИД 11740489 . S2CID 37789883 .
- ^ Ким Э.Дж., Пак Дж.С., Ум С.Дж. (август 2002 г.). «Идентификация и характеристика HIPK2, взаимодействующего с p73 и модулирующего функции семейства p53 in vivo» . Журнал биологической химии . 277 (35): 32020–8. дои : 10.1074/jbc.M200153200 . ПМИД 11925430 .
- ^ Имамура Т., Изуми Х., Нагатани Г. и др. (март 2001 г.). «Взаимодействие с р53 усиливает связывание модифицированной цисплатином ДНК высокомобильным белком группы 1» . Журнал биологической химии . 276 (10): 7534–40. дои : 10.1074/jbc.M008143200 . ПМИД 11106654 .
- ^ Динтильяк А., Бернюес Дж. (март 2002 г.). «HMGB1 взаимодействует со многими явно неродственными белками, распознавая короткие аминокислотные последовательности» . Журнал биологической химии . 277 (9): 7021–8. дои : 10.1074/jbc.M108417200 . hdl : 10261/112516 . ПМИД 11748221 .
- ^ Вадхва Р., Ягучи Т., Хасан М.К. и др. (апрель 2002 г.). «Член семейства Hsp70, mot-2/mthsp70/GRP75, связывается с доменом цитоплазматической секвестрации белка p53». Экспериментальные исследования клеток . 274 (2): 246–53. дои : 10.1006/excr.2002.5468 . ПМИД 11900485 .
- ^ Стеффан Дж.С., Казанцев А., Спасич-Бошович О. и др. (июнь 2000 г.). «Белок болезни Хантингтона взаимодействует с p53 и CREB-связывающим белком и подавляет транскрипцию» . Труды Национальной академии наук Соединенных Штатов Америки . 97 (12): 6763–8. Бибкод : 2000PNAS...97.6763S . дои : 10.1073/pnas.100110097 . ЧВК 18731 . ПМИД 10823891 .
- ^ Люнг К.М., По Л.С., Цанг Ф.К. и др. (сентябрь 2002 г.). «Кандидатный супрессор опухоли ING1b может стабилизировать р53, нарушая регуляцию р53 с помощью MDM2». Исследования рака . 62 (17): 4890–3. ПМИД 12208736 .
- ^ Гаркавцев И., Григорян И.А., Оссовская В.С. и др. (январь 1998 г.). «Кандидатный супрессор опухоли p33ING1 взаимодействует с p53 в контроле роста клеток». Природа . 391 (6664): 295–8. Бибкод : 1998Natur.391..295G . дои : 10.1038/34675 . ПМИД 9440695 . S2CID 4429461 .
- ^ Перейти обратно: а б Шисеки М., Нагасима М., Педо Р.М. и др. (май 2003 г.). «p29ING4 и p28ING5 связываются с p53 и p300 и усиливают активность p53». Исследования рака . 63 (10): 2373–8. ПМИД 12750254 .
- ^ Цай К.В., Ценг ХК, Лин В.К. (октябрь 2008 г.). «Два события колебательного сплайсинга влияют на субъядерную локализацию и деградацию белка ING4». Экспериментальные исследования клеток . 314 (17): 3130–41. дои : 10.1016/j.yexcr.2008.08.002 . ПМИД 18775696 .
- ^ Чанг Н.С. (март 2002 г.). «Неанкириновый С-конец икаппы-бальфы физически взаимодействует с p53 in vivo и диссоциирует в ответ на апоптотический стресс, гипоксию, повреждение ДНК и подавление роста, опосредованное трансформирующим фактором роста бета 1» . Журнал биологической химии . 277 (12): 10323–31. дои : 10.1074/jbc.M106607200 . ПМИД 11799106 .
- ^ Перейти обратно: а б Курки С., Латонен Л., Лайхо М. (октябрь 2003 г.). «Клеточный стресс и повреждение ДНК вызывают различные во времени комплексы Mdm2, p53 и PML и специфичную для повреждения ядерную релокализацию» . Журнал клеточной науки . 116 (Часть 19): 3917–25. дои : 10.1242/jcs.00714 . ПМИД 12915590 .
- ^ Перейти обратно: а б Фриман Д.Д., Ли А.Г., Вэй Дж. и др. (февраль 2003 г.). «Супрессор опухоли PTEN регулирует уровни и активность белка p53 посредством фосфатазозависимых и независимых механизмов» . Раковая клетка . 3 (2): 117–30. дои : 10.1016/S1535-6108(03)00021-7 . ПМИД 12620407 .
- ^ Перейти обратно: а б Чжан Ю, Сюн Ю, Ярбро В.Г. (март 1998 г.). «ARF способствует деградации MDM2 и стабилизирует p53: делеция локуса ARF-INK4a нарушает пути подавления опухоли как Rb, так и p53» . Клетка . 92 (6): 725–34. дои : 10.1016/S0092-8674(00)81401-4 . ПМИД 9529249 .
- ^ Бадчионг Дж.К., Хаас А.Л. (декабрь 2002 г.). «MdmX представляет собой убиквитинлигазу RING-пальца, способную синергически усиливать убиквитинирование Mdm2» . Журнал биологической химии . 277 (51): 49668–75. дои : 10.1074/jbc.M208593200 . ПМИД 12393902 .
- ^ Шварц А., Базуин М., Деккер П. и др. (июль 1997 г.). «Выделение и идентификация человеческого гомолога нового p53-связывающего белка, Mdmx» (PDF) . Геномика . 43 (1): 34–42. дои : 10.1006/geno.1997.4775 . hdl : 2066/142231 . ПМИД 9226370 . S2CID 11794685 .
- ^ Фраде Р., Бальбо М., Барель М. (декабрь 2000 г.). «RB18A, ген которого локализован на хромосоме 17q12-q21.1, регулирует трансактивирующую активность р53 in vivo». Исследования рака . 60 (23): 6585–9. ПМИД 11118038 .
- ^ Дране П., Барель М., Бальбо М. и др. (декабрь 1997 г.). «Идентификация RB18A, нового регуляторного белка р53 массой 205 кДа, который разделяет антигенные и функциональные свойства с р53» . Онкоген . 15 (25): 3013–24. дои : 10.1038/sj.onc.1201492 . ПМИД 9444950 .
- ^ Ху MC, Цю WR, Ван Ю.П. (ноябрь 1997 г.). «JNK1, JNK2 и JNK3 представляют собой N-концевые сериновые 34 киназы p53» . Онкоген . 15 (19): 2277–87. дои : 10.1038/sj.onc.1201401 . ПМИД 9393873 .
- ^ Лин Ю., Хохлачев А., Фигейс Д. и др. (декабрь 2002 г.). «Связанный со смертью белок 4 связывает MST1 и усиливает апоптоз, индуцированный MST1» . Журнал биологической химии . 277 (50): 47991–8001. дои : 10.1074/jbc.M202630200 . ПМИД 12384512 .
- ^ Таниура Х., Мацумото К., Ёсикава К. (июнь 1999 г.). «Физические и функциональные взаимодействия супрессора роста нейронов некдина с p53» . Журнал биологической химии . 274 (23): 16242–8. дои : 10.1074/jbc.274.23.16242 . ПМИД 10347180 .
- ^ Дэниели Ю., Димитрова Д.Д., Боровец Ю.А. (август 2002 г.). «Стресс-зависимая мобилизация нуклеолина, опосредованная образованием комплекса p53-нуклеолин» . Молекулярная и клеточная биология . 22 (16): 6014–22. дои : 10.1128/MCB.22.16.6014-6022.2002 . ПМК 133981 . ПМИД 12138209 .
- ^ Колалука И.Н., Тосони Д., Нусифоро П. и др. (январь 2008 г.). «NUMB контролирует активность опухолевого супрессора p53». Природа . 451 (7174): 76–80. Бибкод : 2008Natur.451...76C . дои : 10.1038/nature06412 . ПМИД 18172499 . S2CID 4431258 .
- ^ Перейти обратно: а б с Чой М.К., Мовасса М., Сиггенс Л. и др. (июнь 2010 г.). «Высокопроизводительное секвенирование идентифицирует STAT3 как ДНК-ассоциированный фактор экспрессии генов, зависимой от комплекса p53-NF-kappaB, при сердечной недостаточности у человека» . Геномная медицина . 2 (6): 37. дои : 10,1186/гм158 . ПМК 2905097 . ПМИД 20546595 .
- ^ Перейти обратно: а б Чжан Ю., Вольф Г.В., Бхат К. и др. (декабрь 2003 г.). «Рибосомальный белок L11 отрицательно регулирует онкопротеин MDM2 и опосредует p53-зависимый путь рибосомально-стрессовой контрольной точки» . Молекулярная и клеточная биология . 23 (23): 8902–12. дои : 10.1128/MCB.23.23.8902-8912.2003 . ПМК 262682 . ПМИД 14612427 .
- ^ Николаев А.Ю., Ли М., Пушкаш Н. и др. (январь 2003 г.). «Parc: цитоплазматический якорь р53» . Клетка . 112 (1): 29–40. дои : 10.1016/S0092-8674(02)01255-2 . ПМИД 12526791 .
- ^ Маланга М., Плешке Дж.М., Клечковска Х.Е. и др. (май 1998 г.). «Поли(АДФ-рибоза) связывается со специфическими доменами р53 и изменяет его функции связывания ДНК» . Журнал биологической химии . 273 (19): 11839–43. дои : 10.1074/jbc.273.19.11839 . ПМИД 9565608 .
- ^ Кахьо Т., Нисида Т., Ясуда Х. (сентябрь 2001 г.). «Участие PIAS1 в сумойлировании опухолевого супрессора p53» . Молекулярная клетка . 8 (3): 713–8. дои : 10.1016/S1097-2765(01)00349-5 . ПМИД 11583632 .
- ^ Вульф Г.М., Лиу Ю.К., Рио А. и др. (декабрь 2002 г.). «Роль Pin1 в регуляции стабильности p53 и трансактивации p21, а также контрольных точек клеточного цикла в ответ на повреждение ДНК» . Журнал биологической химии . 277 (50): 47976–9. дои : 10.1074/jbc.C200538200 . ПМИД 12388558 .
- ^ Закки П., Гостисса М., Учида Т. и др. (октябрь 2002 г.). «Пролилизомераза Pin1 раскрывает механизм контроля функций р53 после генотоксических инсультов». Природа . 419 (6909): 853–7. Бибкод : 2002Natur.419..853Z . дои : 10.1038/nature01120 . ПМИД 12397362 . S2CID 4311658 .
- ^ Хуанг С.М., Шёнталь А.Х., Столлкап MR (апрель 2001 г.). «Усиление р53-зависимой активации гена с помощью транскрипционного коактиватора Zac1». Онкоген . 20 (17): 2134–43. дои : 10.1038/sj.onc.1204298 . ПМИД 11360197 . S2CID 21331603 .
- ^ Се С., Ву Х, Ван Ц и др. (ноябрь 2001 г.). «Plk3 функционально связывает повреждение ДНК с остановкой клеточного цикла и апоптозом, по крайней мере частично, через путь p53» . Журнал биологической химии . 276 (46): 43305–12. дои : 10.1074/jbc.M106050200 . ПМИД 11551930 .
- ^ Бахасси Э.М., Конн К.В., Майер Д.Л. и др. (сентябрь 2002 г.). «Поло-подобная киназа 3 млекопитающих (Plk3) представляет собой многофункциональный белок, участвующий в путях реакции на стресс». Онкоген . 21 (43): 6633–40. дои : 10.1038/sj.onc.1205850 . ПМИД 12242661 . S2CID 24106070 .
- ^ Саймонс А., Меламед-Бессудо С., Волкович Р. и др. (январь 1997 г.). «ПАКТ: клонирование и характеристика клеточного белка, связывающего р53, который взаимодействует с Rb» . Онкоген . 14 (2): 145–55. дои : 10.1038/sj.onc.1200825 . ПМИД 9010216 .
- ^ Фусаро Г., Дасгупта П., Растоги С. и др. (ноябрь 2003 г.). «Прогибин индуцирует транскрипционную активность р53 и экспортируется из ядра при апоптотической передаче сигналов» . Журнал биологической химии . 278 (48): 47853–61. дои : 10.1074/jbc.M305171200 . ПМИД 14500729 .
- ^ Фогал В., Гостисса М., Сэнди П. и др. (ноябрь 2000 г.). «Регуляция активности р53 в ядерных тельцах с помощью специфической изоформы PML» . Журнал ЭМБО . 19 (22): 6185–95. дои : 10.1093/emboj/19.22.6185 . ПМК 305840 . ПМИД 11080164 .
- ^ Го А., Саломони П., Луо Дж. и др. (октябрь 2000 г.). «Функция PML в p53-зависимом апоптозе». Природная клеточная биология . 2 (10): 730–6. дои : 10.1038/35036365 . ПМИД 11025664 . S2CID 13480833 .
- ^ Перейти обратно: а б Чжан З, Чжан Р (март 2008 г.). «Протеасомный активатор гамма PA28 регулирует р53, усиливая его деградацию, опосредованную MDM2» . Журнал ЭМБО . 27 (6): 852–64. дои : 10.1038/emboj.2008.25 . ПМК 2265109 . ПМИД 18309296 .
- ^ Лим С.Т., Чен С.Л., Лим Ю. и др. (январь 2008 г.). «Ядерный FAK способствует пролиферации и выживанию клеток за счет деградации p53, усиленной FERM» . Молекулярная клетка . 29 (1): 9–22. doi : 10.1016/j.molcel.2007.11.031 . ПМК 2234035 . ПМИД 18206965 .
- ^ Бернал Дж.А., Луна Р., Эспина А. и др. (октябрь 2002 г.). «Человеческий секурин взаимодействует с p53 и модулирует p53-опосредованную транскрипционную активность и апоптоз». Природная генетика . 32 (2): 306–11. дои : 10.1038/ng997 . ПМИД 12355087 . S2CID 1770399 .
- ^ Стюрцбехер Х.В., Донзельманн Б., Хеннинг В. и др. (апрель 1996 г.). «p53 напрямую связан с процессами гомологичной рекомбинации посредством взаимодействия белков RAD51/RecA» . Журнал ЭМБО . 15 (8): 1992–2002. дои : 10.1002/j.1460-2075.1996.tb00550.x . ПМК 450118 . ПМИД 8617246 .
- ^ Буххоп С., Гибсон М.К., Ван XW и др. (октябрь 1997 г.). «Взаимодействие р53 с белком Rad51 человека» . Исследования нуклеиновых кислот . 25 (19): 3868–74. дои : 10.1093/нар/25.19.3868 . ПМК 146972 . ПМИД 9380510 .
- ^ Ленг Р.П., Лин Ю., Ма В. и др. (март 2003 г.). «Pirh2, p53-индуцированная убиквитин-белковая лигаза, способствует деградации p53» . Клетка . 112 (6): 779–91. дои : 10.1016/S0092-8674(03)00193-4 . ПМИД 12654245 .
- ^ Шэн Ю., Лайстер Р.К., Лемак А. и др. (декабрь 2008 г.). «Молекулярные основы убиквитилирования p53, опосредованного Pirh2» . Структурная и молекулярная биология природы . 15 (12): 1334–42. дои : 10.1038/nsmb.1521 . ПМК 4075976 . ПМИД 19043414 .
- ^ Романова Л.Ю., Виллерс Х., Благосклонный М.В. и др. (декабрь 2004 г.). «Взаимодействие р53 с репликационным белком А опосредует подавление гомологичной рекомбинации». Онкоген . 23 (56): 9025–33. дои : 10.1038/sj.onc.1207982 . ПМИД 15489903 . S2CID 23482723 .
- ^ Рива Ф., Зуко В., Винк А.А. и др. (декабрь 2001 г.). «Индуцированное УФ-излучением разрез ДНК и рекрутирование ядерного антигена пролиферирующих клеток в места репарации происходят независимо от взаимодействия белка А репликации p53 в клетках p53 дикого типа и мутантных клетках карциномы яичника». Канцерогенез . 22 (12): 1971–8. doi : 10.1093/carcin/22.12.1971 . ПМИД 11751427 .
- ^ Линь Дж., Ян К., Ян З. и др. (август 2004 г.). «Ингибирование S100B восстанавливает уровни p53 в клетках первичного злокачественного рака меланомы» . Журнал биологической химии . 279 (32): 34071–7. дои : 10.1074/jbc.M405419200 . ПМИД 15178678 .
- ^ Перейти обратно: а б Минти А., Дюмон Х., Кагад М. и др. (ноябрь 2000 г.). «Ковалентная модификация p73альфа с помощью SUMO-1. Двухгибридный скрининг с помощью p73 идентифицирует новые белки, взаимодействующие с SUMO-1, и мотив взаимодействия SUMO-1» . Журнал биологической химии . 275 (46): 36316–23. дои : 10.1074/jbc.M004293200 . ПМИД 10961991 .
- ^ Перейти обратно: а б Иванчук С.М., Мондал С., Рутка Дж.Т. (июнь 2008 г.). «p14ARF взаимодействует с DAXX: влияние на HDM2 и p53» . Клеточный цикл . 7 (12): 1836–50. дои : 10.4161/cc.7.12.6025 . ПМИД 18583933 .
- ^ Перейти обратно: а б Ли Д., Ким Дж.В., Со Т. и др. (июнь 2002 г.). «Комплекс SWI/SNF взаимодействует с опухолевым супрессором р53 и необходим для активации р53-опосредованной транскрипции» . Журнал биологической химии . 277 (25): 22330–7. дои : 10.1074/jbc.M111987200 . ПМИД 11950834 .
- ^ Янг Пи Джей, Дэй ПМ, Чжоу Дж и др. (январь 2002 г.). «Прямое взаимодействие между белком выживающего мотонейрона и р53 и его связь со спинальной мышечной атрофией» . Журнал биологической химии . 277 (4): 2852–9. дои : 10.1074/jbc.M108769200 . ПМИД 11704667 .
- ^ Сето Э., Ушева А., Замбетти Г.П. и др. (декабрь 1992 г.). «Р53 дикого типа связывается с ТАТА-связывающим белком и подавляет транскрипцию» . Труды Национальной академии наук Соединенных Штатов Америки . 89 (24): 12028–32. Бибкод : 1992PNAS...8912028S . дои : 10.1073/pnas.89.24.12028 . ПМК 50691 . ПМИД 1465435 .
- ^ Цвекл А., Кашанчи Ф., Брейди Дж.Н. и др. (июнь 1999 г.). «Взаимодействие Pax-6 с белком, связывающим ТАТА-бокс, и белком ретинобластомы». Исследовательская офтальмология и визуальные науки . 40 (7): 1343–50. ПМИД 10359315 .
- ^ Макферсон Л.А., Локтев А.В., Вейгель Р.Дж. (ноябрь 2002 г.). «Опухолесупрессорная активность AP2альфа, опосредованная прямым взаимодействием с p53» . Журнал биологической химии . 277 (47): 45028–33. дои : 10.1074/jbc.M208924200 . ПМИД 12226108 .
- ^ Соренсен Т.С., Гирлинг Р., Ли К.В. и др. (октябрь 1996 г.). «Функциональное взаимодействие DP-1 и p53» . Молекулярная и клеточная биология . 16 (10): 5888–95. дои : 10.1128/mcb.16.10.5888 . ПМК 231590 . ПМИД 8816502 .
- ^ Грин Д.Р., Чипук Дж.Э. (июль 2006 г.). «p53 и метаболизм: внутри TIGAR» . Клетка . 126 (1): 30–2. дои : 10.1016/j.cell.2006.06.032 . ПМИД 16839873 .
- ^ Гобер С., Складановски А., Ларсен А.К. (август 1999 г.). «Взаимодействие между р53 и ДНК-топоизомеразой I регулируется по-разному в клетках с диким типом и мутантным р53» . Труды Национальной академии наук Соединенных Штатов Америки . 96 (18): 10355–60. Бибкод : 1999PNAS...9610355G . дои : 10.1073/pnas.96.18.10355 . ЧВК 17892 . ПМИД 10468612 .
- ^ Мао Ю., Мель И.Р., Мюллер М.Т. (февраль 2002 г.). «Субъядерное распределение топоизомеразы I связано с продолжающейся транскрипцией и статусом р53» . Труды Национальной академии наук Соединенных Штатов Америки . 99 (3): 1235–40. Бибкод : 2002PNAS...99.1235M . дои : 10.1073/pnas.022631899 . ПМК 122173 . ПМИД 11805286 .
- ^ Перейти обратно: а б Коуэлл И.Г., Окороков А.Л., Каттс С.А. и др. (февраль 2000 г.). «Человеческие топоизомеразы IIальфа и IIбета взаимодействуют с С-концевой областью р53». Экспериментальные исследования клеток . 255 (1): 86–94. дои : 10.1006/excr.1999.4772 . PMID 10666337 .
- ^ Derbyshire DJ, Basu BP, Serpell LC и др. (июль 2002 г.). «Кристаллическая структура доменов BRCT человека 53BP1, связанных с опухолевым супрессором p53» . Журнал ЭМБО . 21 (14): 3863–72. дои : 10.1093/emboj/cdf383 . ПМК 126127 . ПМИД 12110597 .
- ^ Экблад CM, Фридлер А, Вепринцев Д и др. (март 2004 г.). «Сравнение BRCT-доменов BRCA1 и 53BP1: биофизический анализ» . Белковая наука . 13 (3): 617–25. дои : 10.1110/ps.03461404 . ПМК 2286730 . ПМИД 14978302 .
- ^ Ло К.В., Кан Х.М., Чан Л.Н. и др. (март 2005 г.). «Легкая цепь динеина массой 8 кДа связывается с р53-связывающим белком 1 и опосредует накопление р53 в ядре, вызванное повреждением ДНК» . Журнал биологической химии . 280 (9): 8172–9. дои : 10.1074/jbc.M411408200 . ПМИД 15611139 .
- ^ Джу В.С., Джеффри П.Д., Кантор С.Б. и др. (март 2002 г.). «Структура региона BRCT 53BP1, связанного с p53, и ее сравнение со структурой BRCT Brca1» . Гены и развитие . 16 (5): 583–93. дои : 10.1101/gad.959202 . ПМК 155350 . ПМИД 11877378 .
- ^ Derbyshire DJ, Basu BP, Date T и др. (октябрь 2002 г.). «Очистка, кристаллизация и предварительный рентгеновский анализ доменов BRCT человеческого 53BP1, связанных с опухолевым супрессором p53». Акта Кристаллографика Д. 58 (Часть 10, Часть 2): 1826–9. Бибкод : 2002AcCrD..58.1826D . дои : 10.1107/S0907444902010910 . ПМИД 12351827 .
- ^ Перейти обратно: а б Ивабучи К., Бартель П.Л., Ли Б. и др. (июнь 1994 г.). «Два клеточных белка, которые связываются с р53 дикого типа, но не с мутантным» . Труды Национальной академии наук Соединенных Штатов Америки . 91 (13): 6098–102. Бибкод : 1994PNAS...91.6098I . дои : 10.1073/pnas.91.13.6098 . ПМЦ 44145 . ПМИД 8016121 .
- ^ Наумовски Л., Клири М.Л. (июль 1996 г.). «Р53-связывающий белок 53BP2 также взаимодействует с Bc12 и препятствует развитию клеточного цикла на уровне G2/M» . Молекулярная и клеточная биология . 16 (7): 3884–92. дои : 10.1128/MCB.16.7.3884 . ПМК 231385 . ПМИД 8668206 .
- ^ Томасини Р., Самир А.А., Кэрриер А и др. (сентябрь 2003 г.). «TP53INP1 и протеинкиназа-2, взаимодействующая с гомеодоменом (HIPK2), являются партнерами в регуляции активности p53» . Журнал биологической химии . 278 (39): 37722–9. дои : 10.1074/jbc.M301979200 . ПМИД 12851404 .
- ^ Окамура С., Аракава Х., Танака Т. и др. (июль 2001 г.). «p53DINP1, ген, индуцируемый p53, регулирует p53-зависимый апоптоз» . Молекулярная клетка . 8 (1): 85–94. дои : 10.1016/S1097-2765(01)00284-2 . ПМИД 11511362 .
- ^ Ли Л., Ляо Дж., Руланд Дж. и др. (февраль 2001 г.). «Регуляторный контур TSG101/MDM2 модулирует деградацию MDM2 и контроль обратной связи MDM2/p53» . Труды Национальной академии наук Соединенных Штатов Америки . 98 (4): 1619–24. Бибкод : 2001PNAS...98.1619L . дои : 10.1073/pnas.98.4.1619 . ПМК 29306 . ПМИД 11172000 .
- ^ Ляхович А, депутат Шехара (апрель 2003 г.). «Образование супрамолекулярного комплекса между Rad6 и белками пути p53 во время ответа, вызванного повреждением ДНК» . Молекулярная и клеточная биология . 23 (7): 2463–75. дои : 10.1128/MCB.23.7.2463-2475.2003 . ПМК 150718 . ПМИД 12640129 .
- ^ Шен З., Пардингтон-Пуртимун П.Е., Комо Дж.К. и др. (октябрь 1996 г.). «Ассоциации UBE2I с белками RAD52, UBL1, p53 и RAD51 в двугибридной системе дрожжей» . Геномика . 37 (2): 183–6. дои : 10.1006/geno.1996.0540 . ПМИД 8921390 .
- ^ Бернье-Вилламор В., Сэмпсон Д.А., Матунис М.Дж. и др. (февраль 2002 г.). «Структурная основа E2-опосредованной конъюгации SUMO, выявленная комплексом между убиквитин-конъюгирующим ферментом Ubc9 и RanGAP1» . Клетка . 108 (3): 345–56. дои : 10.1016/S0092-8674(02)00630-X . ПМИД 11853669 .
- ^ Сехат Б., Андерссон С., Гирнита Л. и др. (июль 2008 г.). «Идентификация c-Cbl как новой лигазы для рецептора инсулиноподобного фактора роста-I с отличной от Mdm2 ролью в убиквитинировании и эндоцитозе рецептора». Исследования рака . 68 (14): 5669–77. дои : 10.1158/0008-5472.CAN-07-6364 . ПМИД 18632619 .
- ^ Сонг М.С., Сонг С.Дж., Ким С.Ю. и др. (июль 2008 г.). «Супрессор опухоли RASSF1A способствует самоубиквитинированию MDM2, разрушая комплекс MDM2-DAXX-HAUSP» . Журнал ЭМБО . 27 (13): 1863–74. дои : 10.1038/emboj.2008.115 . ПМЦ 2486425 . ПМИД 18566590 .
- ^ Ян В., Дикер Д.Т., Чен Дж. и др. (март 2008 г.). «CARPs усиливают оборот p53, разрушая 14-3-3сигму и стабилизируя MDM2» . Клеточный цикл . 7 (5): 670–82. дои : 10.4161/cc.7.5.5701 . ПМИД 18382127 .
- ^ Абэ Ю., Ода-Сато Э., Тобиуме К. и др. (март 2008 г.). «Передача сигналов Hedgehog подавляет подавление опухоли, опосредованное р53, путем активации Mdm2» . Труды Национальной академии наук Соединенных Штатов Америки . 105 (12): 4838–43. Бибкод : 2008PNAS..105.4838A . дои : 10.1073/pnas.0712216105 . ПМК 2290789 . ПМИД 18359851 .
- ^ Домесен С., Кеппель М., Доббельштейн М. (январь 2008 г.). «Специфическое ингибирование неддилирования, опосредованного Mdm2, с помощью Tip60». Клеточный цикл . 7 (2): 222–31. дои : 10.4161/cc.7.2.5185 . ПМИД 18264029 . S2CID 8023403 .
- ^ Ли М., Чен Д., Шайло А. и др. (апрель 2002 г.). «Дубиквитинирование p53 с помощью HAUSP является важным путем стабилизации p53». Природа . 416 (6881): 648–53. Бибкод : 2002Natur.416..648L . дои : 10.1038/nature737 . ПМИД 11923872 . S2CID 4389394 .
- ^ Брош Р.М., Кармакар П., Соммерс Дж.А. и др. (сентябрь 2001 г.). «p53 модулирует экзонуклеазную активность белка синдрома Вернера» . Журнал биологической химии . 276 (37): 35093–102. дои : 10.1074/jbc.M103332200 . ПМИД 11427532 .
- ^ Чанг Н.С., Пратт Н., Хит Дж. и др. (февраль 2001 г.). «Индукция гиалуронидазой оксидоредуктазы, содержащей домен WW, которая усиливает цитотоксичность фактора некроза опухоли» . Журнал биологической химии . 276 (5): 3361–70. дои : 10.1074/jbc.M007140200 . ПМИД 11058590 .
- ^ Окамото Т., Изуми Х., Имамура Т. и др. (декабрь 2000 г.). «Прямое взаимодействие р53 с белком, связывающим Y-бокс, YB-1: механизм регуляции экспрессии генов человека». Онкоген . 19 (54): 6194–202. дои : 10.1038/sj.onc.1204029 . ПМИД 11175333 . S2CID 19222684 .
- ^ Келли К.Д., Миллер К.Р., Тодд А. и др. (май 2010 г.). «YPEL3, ген, регулируемый р53, который вызывает клеточное старение» . Исследования рака . 70 (9): 3566–75. дои : 10.1158/0008-5472.CAN-09-3219 . ПМК 2862112 . ПМИД 20388804 .
- ^ Уотерман М.Дж., Ставриди Э.С., Уотерман Дж.Л. и др. (июнь 1998 г.). «АТМ-зависимая активация р53 включает дефосфорилирование и ассоциацию с белками 14-3-3». Природная генетика . 19 (2): 175–8. дои : 10.1038/542 . ПМИД 9620776 . S2CID 26600934 .
- ^ Лю Дж., Гроган Л., Нау М.М. и др. (апрель 2001 г.). «Физическое взаимодействие между p53 и геном первичного ответа Egr-1». Международный журнал онкологии . 18 (4): 863–70. дои : 10.3892/ijo.18.4.863 . ПМИД 11251186 .
- ^ Бай Л., торговец JL (июль 2001 г.). «ZBP-89 способствует остановке роста за счет стабилизации р53» . Молекулярная и клеточная биология . 21 (14): 4670–83. дои : 10.1128/MCB.21.14.4670-4683.2001 . ПМК 87140 . ПМИД 11416144 .
- ^ Ямакучи М., Ловенштейн CJ (март 2009 г.). «МиР-34, SIRT1 и p53: петля обратной связи» . Клеточный цикл . 8 (5): 712–5. дои : 10.4161/cc.8.5.7753 . ПМИД 19221490 .
- ^ Ван Ю, Чжан Дж, Ли Дж и др. (май 2019 г.). «CircRNA_014511 влияет на радиочувствительность мезенхимальных стволовых клеток костного мозга путем связывания с миР-29b-2-5p» . Боснийский журнал фундаментальных медицинских наук . 19 (2): 155–163. дои : 10.17305/bjbms.2019.3935 . ПМЦ 6535393 . ПМИД 30640591 .
Внешние ссылки
[ редактировать ]- «База знаний p53» . Лейн Групп в Институте молекулярной и клеточной биологии (IMCB), Сингапур. Архивировано из оригинала 3 января 2006 г. Проверено 6 апреля 2008 г.
- Запись GeneReviews/NCBI/NIH/UW о синдроме Ли-Фраумени
- ОПУХОЛЕВОЙ БЕЛОК p53 @ OMIM
- p53 восстановление функции
- стр53 @ Атлас генетики и цитогенетики в онкологии и гематологии
- TP53 Джин @ GeneCards
- p53 Новости предоставлены научной организацией
- Гудсел Д.С. (1 июля 2002 г.). «Супрессор опухоли р53» . Молекула месяца . Банк данных белков RCSB . Проверено 6 апреля 2008 г.
- Сусси Т. «Веб-сайт p53» . Проверено 6 апреля 2008 г.
- Living LFS Некоммерческая организация поддержки пациентов с синдромом Ли-Фраумени.
- The George Pantziarka TP53 Trust Группа поддержки из Великобритании для людей с синдромом Ли-Фраумени или другими расстройствами, связанными с TP53.
- База данных соматических мутаций IARC TP53, которую ведет Магали Оливье в IARC, Лион.
- PDBe-KB предоставляет обзор всей информации о структуре, доступной в PDB для Human P53.
- научная анимация конформационные изменения р53 при связывании с ДНК
галерея PDB |
|---|