синдром Робертса
| синдром Робертса | |
|---|---|
| Другие имена | Синдром гипомелии-гипотрихоза-лицевой гемангиомы, синдром SC (ранее считавшийся совершенно отдельным заболеванием), псевдоталидомидный синдром, синдром фокомелии Робертса-SC, синдром фокомелии SC, синдром Аппельта-Геркена-Ленца, RBS, псевдоталидомидный синдром SC и тетрафокомелия. синдром расщелины неба. [ 1 ] [ 2 ] [ 3 ] [ 4 ] |
 | |
| Специальность | Медицинская генетика |
| Назван в честь | Джон Бингэм Робертс |
Синдром Робертса , или иногда называемый псевдоталидомидным синдромом , представляет собой чрезвычайно редкое аутосомно-рецессивное генетическое заболевание, которое характеризуется легкой или тяжелой внутриутробной задержкой или нарушением деления клеток , что приводит к порокам развития костей черепа, лица, рук и ног.
Это вызвано мутацией гена ESCO2 . Это одно из самых редких аутосомно-рецессивных заболеваний, которым страдают около 150 известных людей. Мутация приводит к тому, что деление клеток происходит медленно или неравномерно, а клетки с аномальным генетическим содержимым умирают.
Синдром Робертса может поражать как мужчин, так и женщин. Хотя заболевание встречается редко, группа больных разнообразна. Уровень смертности среди тяжело пораженных людей высок. Синдром назван в честь американского хирурга и врача Джона Бингама Робертса (1852–1924), который впервые описал его в 1919 году.
Симптомы и признаки
[ редактировать ]Ниже приводится список симптомов, связанных с синдромом Робертса: [ нужна ссылка ]
- Двусторонняя симметричная тетрафокомелия – врожденный дефект, при котором кисти и стопы прикреплены к укороченным рукам и ногам.
- Пренатальная задержка роста

Пример тяжелого пациента с синдромом Робертса - Гипомелия (гипоплазия) – неполное развитие ткани или органа; менее радикальный, чем аплазия, которая вообще не развивается
- Олигодактилия – меньшее, чем обычно, количество пальцев рук или ног.
- Аплазия большого пальца – отсутствие большого пальца.
- Синдактилия – состояние, при котором два или более пальцев рук (или ног) соединяются вместе; соединение может затрагивать кости или только кожу между пальцами.
- Клинодактилия – искривление пятого пальца (мизинца) в сторону четвертого пальца (безымянного пальца) вследствие недоразвития средней кости пятого пальца.
- Контрактуры сгибания локтя/колена – неспособность полностью выпрямить руку или ногу.
- Заячья губа – наличие одной или двух вертикальных трещин верхней губы; может быть с одной стороны (односторонний) или с обеих сторон (двусторонний)
- Расщелина неба – отверстие в нёбе
- Предчелюстное выпячивание – верхняя часть рта выдается дальше нижней части рта.
- Микрогнатия – маленький подбородок
- Микробрахицефалия – размер головы меньше нормального.
- Маларная гипоплазия – недоразвитие скуловых костей.
- Наклонные глазные щели – внешние уголки глаз направлены вниз.
- Глазной гипертелоризм – необычно широко посаженные глаза.
- Экзофтальм – выпячивание глазного яблока.
- Помутнение роговицы – помутнение передней части глаза.
- Гипопластические крылья носа – сужение ноздрей, которое может уменьшить ширину основания носа.
- Нос с клювом - нос с выступающей переносицей, которая придает ему вид изогнутости.
- Пороки развития ушей
- Интеллектуальная инвалидность
- Энцефалоцеле (только в тяжелых случаях) — редкий дефект нервной трубки, характеризующийся мешкообразными выпячиваниями головного мозга.

Смертность высока среди тех, кто серьезно пострадал от синдрома Робертса; однако люди с легким поражением могут дожить до взрослого возраста. [ 1 ] [ 3 ] [ 4 ]
Наследственность
[ редактировать ]
От Херста и Пирсола, 1893 г.
ESCO2 , расположенный на 8-й хромосоме человека , считается геном, ответственным за синдром Робертса. Фактически, ESCO2 — единственный известный ген, который продемонстрировал мутации, вызывающие RBS. был Кроме того, у всех людей, у которых цитогенетически диагностирован синдром Робертса, также были мутации в гене ESCO2. [ 3 ]
Чтобы заразиться синдромом Робертса, ребенок должен унаследовать дефектный ген по аутосомно- рецессивному типу. Другими словами, ребенок должен унаследовать две копии дефектного гена (по одной от каждого родителя). Ген ESCO2 оказывает специфическое влияние на деление клеток у пациентов с синдромом Робертса. При нормальном делении клеток каждая хромосома копируется, а затем прикрепляется к своей вновь образованной копии в центромере (центральной части хромосомы). Однако при делении клеток при синдроме Робертса копии часто не прикрепляются к центромере. В результате хромосомы не выстраиваются должным образом, из-за чего клетка делится очень медленно или даже не делится вообще. Новые клетки обычно будут иметь слишком много или слишком мало хромосом. Нечетное число хромосом приводит к гибели дефектных клеток, что приводит к порокам развития, связанным с синдромом Робертса. [ 1 ]
Многие физические пороки развития, связанные с синдромом Робертса, очень похожи на пороки развития, возникающие у детей, матери которых принимали талидомид во время беременности. Физическое сходство позволяет предположить, что между ESCO2 и талидомидом существует сходная биологическая основа. В результате предполагается, что талидомид влияет на хромосомы и деление клеток аналогично ESCO2. По этой причине синдром Робертса иногда называют синдромом псевдоталидомида. [ нужна ссылка ]
Открытие синдрома
[ редактировать ]Открытие ESCO2 как гена, ответственного за синдром Робертса, было сделано путем изучения образцов пятнадцати семей, страдающих синдромом Робертса. В 1995 году Уго Вега и Мириам Гордилло, два колумбийских генетика, решили полностью понять синдром Робертса. Вега и Гордилло заметили необычно большое количество пациентов с синдромом Робертса в Национальном университете Колумбии . Два колумбийских генетика выследили в общей сложности семь семей с синдромом Робертса недалеко от Боготы и обнаружили, что четыре из семи семей имели общего предка, жившего в 18 веке. Используя эту информацию, Вега и Гордилло смогли точно определить ген, ответственный за синдром Робертса, — ESCO2. [ 5 ]
Диагностика
[ редактировать ]Клинический диагноз
[ редактировать ]Клинический диагноз синдрома Робертса ставят лицам с характерной пренатальной задержкой роста, пороками развития конечностей и черепно-лицевыми аномалиями. Ниже перечислены конкретные характеристики, на которые обращают внимание при клиническом диагнозе. [ нужна ссылка ]
- Пренатальная задержка роста – низкий рост и вес при рождении, которые могут варьироваться от легкой до тяжелой степени.
- Пороки развития конечностей - двусторонняя симметричная тетрафокомелия , олигодактилия, аплазия большого пальца, синдактилия, клинодактилия, сгибательные контрактуры в локтевых и коленных суставах.
- Черепно-лицевые аномалии - двусторонняя расщелина губы и неба, микрогнатия , гипертелоризм , экзофтальм , наклоненные вниз глазные щели , гипоплазия скул, гипопластические крылья носа и пороки развития уха.
Официальный диагноз синдрома Робертса основывается на цитогенетическом тестировании периферической крови. [ 6 ]
Тестирование
[ редактировать ]Цитогенетическое тестирование
[ редактировать ]Цитогенетические препараты, окрашенные методами Гимзы или С-бэндинга, покажут две характерные хромосомные аномалии. Первая хромосомная аномалия называется преждевременным разделением центромер (PCS) и является наиболее вероятным патогенным механизмом синдрома Робертса. У хромосом, имеющих PCS, центромеры будут разделяться во время метафазы, а не анафазы (на одну фазу раньше, чем у нормальных хромосом). Вторая хромосомная аномалия называется отталкиванием гетерохроматина (HR). Хромосомы, имеющие HR, испытывают разделение гетерохроматических областей во время метафазы. Хромосомы с этими двумя аномалиями будут иметь вид «железнодорожного пути» из-за отсутствия первичного сужения и отталкивания в гетерохроматических областях. Гетерохроматиновыми областями являются области вблизи центромер и ядрышковых организаторов. Статус носительства не может быть определен цитогенетическим тестированием. Другие общие результаты цитогенетического тестирования пациентов с синдромом Робертса перечислены ниже.
- Анеуплоидия – появление одной или нескольких дополнительных или недостающих хромосом.
- Микронуклеация – ядро меньше обычного.
- Многодольчатые ядра – ядро имеет более одной доли. [ 6 ]
Генетическое тестирование
[ редактировать ]На данный момент ESCO2 является единственным известным геном, вызывающим мутации синдрома Робертса. Кроме того, у всех людей, у которых с помощью цитогенетических методов был диагностирован синдром Робертса, также были мутации ESCO2. Для подтверждения диагноза синдрома Робертса необходимо обнаружение характерных хромосомных аномалий (PCS и HR) или выявление двух мутаций ESCO2, которые связаны с синдромом Робертса. [ 6 ]
Тестирование на носительство и пренатальная диагностика
[ редактировать ]Тестирование на носительство синдрома Робертса требует предварительного выявления мутации, вызывающей заболевание, в семье. Носителями заболевания являются гетерозиготы из-за аутосомно-рецессивного характера заболевания. Носители также не подвергаются риску заражения синдромом Робертса. Пренатальная диагностика синдрома Робертса требует ультразвукового исследования в сочетании с цитогенетическим тестированием или предварительного выявления мутаций ESCO2, вызывающих заболевание, в семье. [ 6 ]
Генетически связанные заболевания
[ редактировать ]В настоящее время не обнаружено других фенотипов (наблюдаемых проявлений гена) мутаций в гене ESCO2. [ 6 ]
Дифференциальный диагноз
[ редактировать ]При легких пороках развития при дифференциальной диагностике следует учитывать следующие нарушения: [ нужна ссылка ]
- Синдром Баллера-Герольда
- Анемия Фанкони (АФ)
В случаях тяжелых проявлений при дифференциальной диагностике следует учитывать следующие нарушения:
- Синдром тромбоцитопении-отсутствия лучевой кости (TAR)
- Тетра-Амелия, Х-сцепленная
- Тетра-Амелия, аутосомно-рецессивный тип
- Спленогонадальное сращение с дефектами конечностей и микрогнатией
- Синдром ДК-фокомелии
- Синдром Холта-Орама
- Талидомид Эмбриопатия
В случаях сходных цитогенетических данных при дифференциальной диагностике следует учитывать следующие нарушения:
- Синдром Корнелии де Ланге (CdLS)
- Синдром мозаичной пестрой анеуплоидии [ 6 ]
Клиническое описание
[ редактировать ]Мало что известно о естественном течении синдрома Робертса из-за его широкой клинической вариабельности. Прогноз заболевания зависит от пороков развития, поскольку тяжесть пороков развития коррелирует с выживаемостью. Причина смерти большинства смертей от синдрома Робертса неизвестна; однако, как сообщается, пять смертей произошли из-за инфекции. [ нужна ссылка ]
Ниже приведены наблюдения, которые были сделаны у людей с цитогенетическими признаками мутаций PCS/HR или ESCO2: [ нужна ссылка ]
- Симптом пренатальной задержки роста является наиболее частым признаком и может быть от умеренной до тяжелой степени. Постнатальная задержка роста также может быть от умеренной до тяжелой и коррелирует со степенью тяжести пороков развития конечностей и черепно-лицевых суставов.
- При пороках развития конечностей верхние конечности обычно поражаются сильнее, чем нижние. Было много случаев пороков развития только верхних конечностей.
- При пороках развития кисти чаще всего поражается большой палец, затем пятый палец (мизинец). В тяжелых случаях у пациента может быть только три пальца, а в редких случаях - только один.
- При черепно-лицевых пороках развития у людей с легким поражением не наблюдается аномалий неба. Наиболее тяжело пострадает лобно-решетчато-носо-верхнечелюстное энцефалоцеле.
- Тяжесть пороков развития конечностей и черепно-лицевых пороков коррелирует.
- Другие аномалии могут возникать в различных частях тела, в том числе:
- Сердце - дефекты межпредсердной перегородки, дефекты межжелудочковой перегородки, открытый артериальный проток
- Почки - поликистозная почка, подковообразная почка.
- Мужские половые органы – увеличение полового члена, крипторхизм.
- Женские половые органы – увеличенный клитор.
- Волосы : редкие, серебристо-светлые волосы на голове.
- Паралич черепных нервов, болезнь моямоя , инсульт, умственная отсталость [ 3 ]
Уход
[ редактировать ]Лечение синдрома Робертса индивидуализировано и специально направлено на улучшение качества жизни людей, страдающих этим расстройством. Некоторые из возможных методов лечения включают: операцию по поводу расщелины губы и неба, коррекцию аномалий конечностей (также хирургическим путем) и улучшение развития цепкого хвата. [ 3 ]
Распространенность
[ редактировать ]Синдром Робертса — чрезвычайно редкое заболевание, от которого страдают только около 150 человек. Хотя зарегистрировано всего около 150 случаев, затронутая группа весьма разнообразна и распространена по всему миру. Родительское кровное родство (родители являются близкими родственниками) часто встречается при этом генетическом заболевании. Частота носителей синдрома Робертса неизвестна. [ 3 ] [ 4 ]
Номенклатура
[ редактировать ]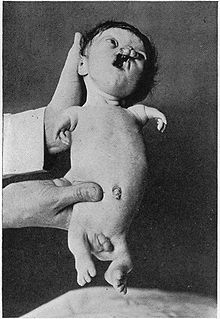
Синдром Робертса назван в честь доктора Джона Бингама Робертса (1852–1924) из Филадельфии, который сообщил о характеристиках заболевания в 1919 году. Робертс сообщил о заболевании, которое характеризовалось фокомелией, расщелиной губы, волчьей пастью и выпячиванием межчелюстной области в трое братьев и сестер итальянской пары, которые приходились двоюродными братьями, что сделало приобретение синдрома Робертса более вероятным для их детей из-за аутосомно-рецессивной природы заболевания. [ 8 ]
Позже, в 1969 г., Дж. Херрманн описал еще один синдром, по характеристикам очень похожий на синдром Робертса. Херрманн назвал бы это расстройство псевдоталидомидным синдромом или синдромом SC (SC - это инициалы фамилий двух семей, которые изучал Херрманн). Сегодня синдром Робертса и синдром псевдоталидомида (синдром SC) считаются одним и тем же заболеванием. [ нужна ссылка ]
Ниже приводится список всех альтернативных названий, которые использовались для синдрома Робертса:
- РБС
- Гипомелия-гипотрихоз-синдром гемангиомы лица
- Синдром СК
- Псевдоталидомидный синдром
- Синдром Робертса-SC Фокомелии
- Синдром SC-фокомелии
- Синдром Аппельта-Геркена-Ленца
- SC псевдоталидомидный синдром
- Синдром тетрафокомелии-волчьей пасти [ 2 ] [ 3 ] [ 4 ]
Ссылки
[ редактировать ]- ^ Jump up to: а б с Куглер, Мэри. «Синдром Робертса: наследственное заболевание вызывает аномальное развитие костей». Архивировано 13 февраля 2009 г. на сайте Wayback Machine About.com: Редкие заболевания. Опубликовано 23 апреля 2005 г. По состоянию на 13 марта 2010 г.
- ^ Jump up to: а б Франке, Ута и Цзинлань Лю. «Синдром Робертса». Национальная организация редких заболеваний. Опубликовано 26 ноября 2008 г.
- ^ Jump up to: а б с д и ж г Гордилло и др. «Синдром Робертса».
- ^ Jump up to: а б с д «Синдром Робертса». Домашний справочник по генетике. 2010. Национальная медицинская библиотека США. 13 марта 2010 г.
- ^ Даунер, Джоанна. «Пятнадцатилетняя охота обнаружила ген, стоящий за синдромом псевдоталидомида». Пресс-релизы. Медицина Джонса Хопкинса. 11 апреля 2005 г.
- ^ Jump up to: а б с д и ж Гордилло, Мириам, Хьюго Вега и Этилин Ван Джабс. «Синдром Робертса». Джин Обзоры . 2009. Вашингтонский университет, Сиэтл. 13 марта 2010 г.
- ^ «Труды Филадельфийской академии хирургии: официальное собрание, состоявшееся 5 мая 1919 г.» . Анналы хирургии . 70 (2): 251–4. 1919. дои : 10.1097/00000658-191908000-00019 . ПМЦ 1410314 . ПМИД 17864157 .
- ^ «ТРАНЗАКЦИИ ФИЛАДЕЛЬФИЙСКОЙ АКАДЕМИИ ХИРУРГИИ Заявленное собрание состоялось 5 мая 1919 года» . Энн. Сург . 70 (2): 251–254. 1919. дои : 10.1097/00000658-191908000-00019 . ПМЦ 1410314 . ПМИД 17864157 .
Дальнейшее чтение
[ редактировать ]- Куглер, Мэри. «Синдром Робертса: наследственное заболевание вызывает аномальное развитие костей». About.com: Редкие заболевания. О . 23 апреля 2005 г.
- Даунер, Джоанна. «Пятнадцатилетняя охота обнаружила ген, стоящий за синдромом псевдоталидомида». Пресс-релизы. Медицина Джонса Хопкинса. 11 апреля 2005 г.
- Франке, Ута и Цзинлань Лю. «Синдром Робертса». Национальная организация редких заболеваний. 26 ноября 2008 г.
- Гордилло, Мириам, Хьюго Вега и Этилин Ван Джабс. «Синдром Робертса». Джин Обзоры. 2009. Вашингтонский университет, Сиэтл. 13 марта 2010 г.
- «Синдром Робертса». Домашний справочник по генетике. 2010. Национальная медицинская библиотека США. По состоянию на 13 марта 2010 г.
- «Синдром Робертса». ВебМД. 2009. 13 марта 2010 г.
- Сильва, Сандра и Филипп Жанти. Синдром Робертса . [1] . 1999. СоноМир. 13 марта 2010 г.
- «Информационная страница NINDS по энцефалоцеле». Национальный институт неврологических расстройств и инсульта. 2007. Национальные институты здравоохранения. 13 марта 2010 г.
Внешние ссылки
[ редактировать ]Хромосомные+нарушения Национальной медицинской библиотеки США по медицинским предметным рубрикам (MeSH)