Кислотная постоянная диссоциация

В химии константа диссоциации кислоты (также известная как константа кислотности , или константа ионизации кислотности ; обозначенная ) количественной мерой силы кислоты является в растворе . Это постоянная равновесия для химической реакции


известный как диссоциация в контексте кислотных реакций . Химические виды HA - это кислота , которая диссоциирует в А − , называется сопряженным основанием кислоты и ион водорода , ЧАС + . [ А ] Говорят, что система находится в равновесии , когда концентрации ее компонентов не изменяются со временем, потому что как вперед, так и назад реакции происходят с той же скоростью. [ 1 ]
Постоянная диссоциации определяется [ B ]
- или по его логарифмической форме
![{\ displayStyle k _ {{\ text {}} = \ mathmr {\ frac {a^{{{{{h^{+}}}}-{[Ha]}},},},},},} ,},},},}, [](https://wikimedia.org/api/rest_v1/media/math/render/svg/7cdd9efda0e3a32060020b5c9e5b2c78981b2a93)
![{\displaystyle \mathrm {p} K_{{\ce {a}}}=-\log _{10}K_{\text{a}}=\log _{10}{\frac {{\ce {[HA]}}}{[{\ce {A^-}}][{\ce {H+}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d7af05bf129db2f9bc618fe809660b6e4ff8dce9)
where quantities in square brackets represent the molar concentrations of the species at equilibrium.[c][2] For example, a hypothetical weak acid having Ka = 10−5, the value of log Ka is the exponent (−5), giving pKa = 5. For acetic acid, Ka = 1.8 x 10−5, so pKa is about 5. A higher Ka corresponds to a stronger acid (an acid that is more dissociated at equilibrium). The form pKa is often used because it provides a convenient logarithmic scale, where a lower pKa corresponds to a stronger acid.
Theoretical background
[edit]The acid dissociation constant for an acid is a direct consequence of the underlying thermodynamics of the dissociation reaction; the pKa value is directly proportional to the standard Gibbs free energy change for the reaction. The value of the pKa changes with temperature and can be understood qualitatively based on Le Châtelier's principle: when the reaction is endothermic, Ka increases and pKa decreases with increasing temperature; the opposite is true for exothermic reactions.
The value of pKa also depends on molecular structure of the acid in many ways. For example, Pauling proposed two rules: one for successive pKa of polyprotic acids (see Polyprotic acids below), and one to estimate the pKa of oxyacids based on the number of =O and −OH groups (see Factors that affect pKa values below). Other structural factors that influence the magnitude of the acid dissociation constant include inductive effects, mesomeric effects, and hydrogen bonding. Hammett type equations have frequently been applied to the estimation of pKa.[3][4]
The quantitative behaviour of acids and bases in solution can be understood only if their pKa values are known. In particular, the pH of a solution can be predicted when the analytical concentration and pKa values of all acids and bases are known; conversely, it is possible to calculate the equilibrium concentration of the acids and bases in solution when the pH is known. These calculations find application in many different areas of chemistry, biology, medicine, and geology. For example, many compounds used for medication are weak acids or bases, and a knowledge of the pKa values, together with the octanol-water partition coefficient, can be used for estimating the extent to which the compound enters the blood stream. Acid dissociation constants are also essential in aquatic chemistry and chemical oceanography, where the acidity of water plays a fundamental role. In living organisms, acid–base homeostasis and enzyme kinetics are dependent on the pKa values of the many acids and bases present in the cell and in the body. In chemistry, a knowledge of pKa values is necessary for the preparation of buffer solutions and is also a prerequisite for a quantitative understanding of the interaction between acids or bases and metal ions to form complexes. Experimentally, pKa values can be determined by potentiometric (pH) titration, but for values of pKa less than about 2 or more than about 11, spectrophotometric or NMR measurements may be required due to practical difficulties with pH measurements.
Definitions
[edit]According to Arrhenius's original molecular definition, an acid is a substance that dissociates in aqueous solution, releasing the hydrogen ion H+ (a proton):[5]

The equilibrium constant for this dissociation reaction is known as a dissociation constant. The liberated proton combines with a water molecule to give a hydronium (or oxonium) ion H3O+ (naked protons do not exist in solution), and so Arrhenius later proposed that the dissociation should be written as an acid–base reaction:

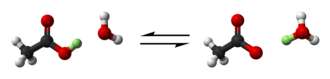
Brønsted and Lowry generalised this further to a proton exchange reaction:[6][7][8]

The acid loses a proton, leaving a conjugate base; the proton is transferred to the base, creating a conjugate acid. For aqueous solutions of an acid HA, the base is water; the conjugate base is A− and the conjugate acid is the hydronium ion. The Brønsted–Lowry definition applies to other solvents, such as dimethyl sulfoxide: the solvent S acts as a base, accepting a proton and forming the conjugate acid SH+.

In solution chemistry, it is common to use H+ as an abbreviation for the solvated hydrogen ion, regardless of the solvent. In aqueous solution H+ denotes a solvated hydronium ion rather than a proton.[9][10]
The designation of an acid or base as "conjugate" depends on the context. The conjugate acid BH+ of a base B dissociates according to

which is the reverse of the equilibrium

The hydroxide ion OH−, a well known base, is here acting as the conjugate base of the acid water. Acids and bases are thus regarded simply as donors and acceptors of protons respectively.
A broader definition of acid dissociation includes hydrolysis, in which protons are produced by the splitting of water molecules. For example, boric acid (B(OH)3) produces H3O+ as if it were a proton donor,[11] but it has been confirmed by Raman spectroscopy that this is due to the hydrolysis equilibrium:[12]

Similarly, metal ion hydrolysis causes ions such as [Al(H2O)6]3+ to behave as weak acids:[13]
![{\displaystyle {\ce {[Al(H2O)6]^3+ + H2O <=> [Al(H2O)5(OH)]^2+ + H3O+}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b1c60923d504a87f8bbd22293ac8eaad8341ea41)
According to Lewis's original definition, an acid is a substance that accepts an electron pair to form a coordinate covalent bond.[14]
Equilibrium constant
[edit]An acid dissociation constant is a particular example of an equilibrium constant. The dissociation of a monoprotic acid, HA, in dilute solution can be written as
The thermodynamic equilibrium constant can be defined by[15]


where represents the activity, at equilibrium, of the chemical species X. is dimensionless since activity is dimensionless. Activities of the products of dissociation are placed in the numerator, activities of the reactants are placed in the denominator. See activity coefficient for a derivation of this expression.


Since activity is the product of concentration and activity coefficient (γ) the definition could also be written as
![{\displaystyle K^{\ominus }={{\frac {[{\ce {A^-}}][{\ce {H+}}]}{{\ce {[HA]}}}}\Gamma },\quad \Gamma ={\frac {\gamma _{{\ce {A^-}}}\ \gamma _{{\ce {H+}}}}{\gamma _{{\ce {HA}}}\ }}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6e9373db7091aeb4f51a26757a677b420f0a8418)
where represents the concentration of HA and is a quotient of activity coefficients.
![{\displaystyle [{\text{HA}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3cfe8305c0735d25de8cef20edf09ef5144d700a)

To avoid the complications involved in using activities, dissociation constants are determined, where possible, in a medium of high ionic strength, that is, under conditions in which can be assumed to be always constant.[15] For example, the medium might be a solution of 0.1 molar (M) sodium nitrate or 3 M potassium perchlorate. With this assumption,
![{\displaystyle K_{\text{a}}={\frac {K^{\ominus }}{\Gamma }}=\mathrm {\frac {[A^{-}][H^{+}]}{[HA]}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/2a5a59c740de89347ec4c96d982292fc05c64b2f)
![{\displaystyle \mathrm {p} K_{{\ce {a}}}=-\log _{10}{\frac {[{\ce {A^-}}][{\ce {H^+}}]}{[{\ce {HA}}]}}=\log _{10}{\frac {{\ce {[HA]}}}{[{\ce {A^-}}][{\ce {H+}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/bed5fbab82167a42994a6d735931d08b06f1e7a5)
is obtained. Note, however, that all published dissociation constant values refer to the specific ionic medium used in their determination and that different values are obtained with different conditions, as shown for acetic acid in the illustration above. When published constants refer to an ionic strength other than the one required for a particular application, they may be adjusted by means of specific ion theory (SIT) and other theories.[16]
Cumulative and stepwise constants
[edit]A cumulative equilibrium constant, denoted by is related to the product of stepwise constants, denoted by For a dibasic acid the relationship between stepwise and overall constants is as follows



![{\displaystyle \beta _{2}={\frac {{\ce {[H2A]}}}{[{\ce {A^2-}}][{\ce {H+}}]^{2}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/08598ffd39aa7af9e4d7ca73764ada00fdc0882f)

Note that in the context of metal-ligand complex formation, the equilibrium constants for the formation of metal complexes are usually defined as association constants. In that case, the equilibrium constants for ligand protonation are also defined as association constants. The numbering of association constants is the reverse of the numbering of dissociation constants; in this example

Association and dissociation constants
[edit]When discussing the properties of acids it is usual to specify equilibrium constants as acid dissociation constants, denoted by Ka, with numerical values given the symbol pKa.
![{\displaystyle K_{\text{dissoc}}={\frac {{\ce {[A- ][H+]}}}{{\ce {[HA]}}}}:\mathrm {p} K_{\text{a}}=-\log K_{\text{dissoc}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c115ab88c5f847b2fe5c3250d9e5c9134d125080)
On the other hand, association constants are used for bases.
![{\displaystyle K_{\text{assoc}}={\frac {{\ce {[HA]}}}{{\ce {[A- ][H+]}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e353beeaa76919ab646b6969e6d30f7e01fe7afa)
However, general purpose computer programs that are used to derive equilibrium constant values from experimental data use association constants for both acids and bases. Because stability constants for a metal-ligand complex are always specified as association constants, ligand protonation must also be specified as an association reaction.[17] The definitions show that the value of an acid dissociation constant is the reciprocal of the value of the corresponding association constant:



Notes
- For a given acid or base in water, pKa + pKb = pKw, the self-ionization constant of water.
- The association constant for the formation of a supramolecular complex may be denoted as Ka; in such cases "a" stands for "association", not "acid".
- For polyprotic acids, the numbering of stepwise association constants is the reverse of the numbering of the dissociation constants. For example, for phosphoric acid (details in the polyprotic acids section below):

Temperature dependence
[edit]All equilibrium constants vary with temperature according to the van 't Hoff equation[18]

is the gas constant and is the absolute temperature. Thus, for exothermic reactions, the standard enthalpy change, , is negative and K decreases with temperature. For endothermic reactions, is positive and K increases with temperature.



The standard enthalpy change for a reaction is itself a function of temperature, according to Kirchhoff's law of thermochemistry:

where is the heat capacity change at constant pressure. In practice may be taken to be constant over a small temperature range.

Dimensionality
[edit]In the equation
![{\displaystyle K_{\mathrm {a} }=\mathrm {\frac {[A^{-}][H^{+}]}{[HA]}} ,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/441ece0dee32e0a14fe14d4b1678785804486a92)
Ka appears to have dimensions of concentration. However, since , the equilibrium constant, , cannot have a physical dimension. This apparent paradox can be resolved in various ways.


- Assume that the quotient of activity coefficients has a numerical value of 1, so that has the same numerical value as the thermodynamic equilibrium constant .
- Express each concentration value as the ratio c/c0, where c0 is the concentration in a [hypothetical] standard state, with a numerical value of 1, by definition.[19]
- Express the concentrations on the mole fraction scale. Since mole fraction has no dimension, the quotient of concentrations will, by definition, be a pure number.
The procedures, (1) and (2), give identical numerical values for an equilibrium constant. Furthermore, since a concentration is simply proportional to mole fraction and density :




and since the molar mass is a constant in dilute solutions, an equilibrium constant value determined using (3) will be simply proportional to the values obtained with (1) and (2).

It is common practice in biochemistry to quote a value with a dimension as, for example, "Ka = 30 mM" in order to indicate the scale, millimolar (mM) or micromolar (μM) of the concentration values used for its calculation.
Strong acids and bases
[edit]An acid is classified as "strong" when the concentration of its undissociated species is too low to be measured.[6] Any aqueous acid with a pKa value of less than 0 is almost completely deprotonated and is considered a strong acid.[20] All such acids transfer their protons to water and form the solvent cation species (H3O+ in aqueous solution) so that they all have essentially the same acidity, a phenomenon known as solvent leveling.[21][22] They are said to be fully dissociated in aqueous solution because the amount of undissociated acid, in equilibrium with the dissociation products, is below the detection limit. Likewise, any aqueous base with an association constant pKb less than about 0, corresponding to pKa greater than about 14, is leveled to OH− and is considered a strong base.[22]
Nitric acid, with a pK value of around −1.7, behaves as a strong acid in aqueous solutions with a pH greater than 1.[23] At lower pH values it behaves as a weak acid.
pKa values for strong acids have been estimated by theoretical means.[24] For example, the pKa value of aqueous HCl has been estimated as −9.3.
Monoprotic acids
[edit]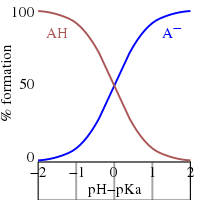
After rearranging the expression defining Ka, and putting pH = −log10[H+], one obtains[25]
![{\displaystyle \mathrm {pH} =\mathrm {p} K_{\text{a}}+\log \mathrm {\frac {[A^{-}]}{[HA]}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/25e874f2b8ea8e4127605788c356393cfd7fff37)
This is the Henderson–Hasselbalch equation, from which the following conclusions can be drawn.
- At half-neutralization the ratio [A−]/[HA] = 1; since log(1) = 0, the pH at half-neutralization is numerically equal to pKa. Conversely, when pH = pKa, the concentration of HA is equal to the concentration of A−.
- The buffer region extends over the approximate range pKa ± 2. Buffering is weak outside the range pKa ± 1. At pH ≤ pKa − 2 the substance is said to be fully protonated and at pH ≥ pKa + 2 it is fully dissociated (deprotonated).
- If the pH is known, the ratio may be calculated. This ratio is independent of the analytical concentration of the acid.
In water, measurable pKa values range from about −2 for a strong acid to about 12 for a very weak acid (or strong base).
A buffer solution of a desired pH can be prepared as a mixture of a weak acid and its conjugate base. In practice, the mixture can be created by dissolving the acid in water, and adding the requisite amount of strong acid or base. When the pKa and analytical concentration of the acid are known, the extent of dissociation and pH of a solution of a monoprotic acid can be easily calculated using an ICE table.
Polyprotic acids
[edit]
A polyprotic acid is a compound which may lose more than 1 proton. Stepwise dissociation constants are each defined for the loss of a single proton. The constant for dissociation of the first proton may be denoted as Ka1 and the constants for dissociation of successive protons as Ka2, etc. Phosphoric acid, H3PO4, is an example of a polyprotic acid as it can lose three protons.
Equilibrium pK definition and value[26]

![{\displaystyle \mathrm {p} K_{{\ce {a1}}}=\log _{10}{\frac {[{\ce {H_3PO_4}}]}{[{\ce {H_2PO_4^{-}}}][{\ce {H^+}}]}}=2.14}](https://wikimedia.org/api/rest_v1/media/math/render/svg/dff6e67381cb8a691b8873fbf884dad30b001352)

![{\displaystyle \mathrm {p} K_{{\ce {a2}}}=\log _{10}{\frac {[{\ce {H_2PO_4^{-}}}]}{[{\ce {HPO_4^{2-}}}][{\ce {H^+}}]}}=7.2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/efe9f5a620a62c8de4a6567f58faf01e66829903)

![{\displaystyle \mathrm {p} K_{{\ce {a3}}}=\log _{10}{\frac {[{\ce {HPO4^2-}}]}{[{\ce {PO4^3-}}][{\ce {H+}}]}}=12.37}](https://wikimedia.org/api/rest_v1/media/math/render/svg/dfac57be595c190a1dc60479c2538e575488de02)
When the difference between successive pK values is about four or more, as in this example, each species may be considered as an acid in its own right;[27] In fact salts of H
2PO−
4 may be crystallised from solution by adjustment of pH to about 5.5 and salts of HPO2−4 may be crystallised from solution by adjustment of pH to about 10. The species distribution diagram shows that the concentrations of the two ions are maximum at pH 5.5 and 10.
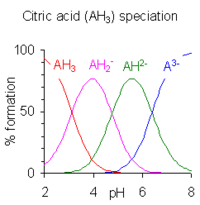
When the difference between successive pK values is less than about four there is overlap between the pH range of existence of the species in equilibrium. The smaller the difference, the more the overlap. The case of citric acid is shown at the right; solutions of citric acid are buffered over the whole range of pH 2.5 to 7.5.
According to Pauling's first rule, successive pK values of a given acid increase (pKa2 > pKa1).[28] For oxyacids with more than one ionizable hydrogen on the same atom, the pKa values often increase by about 5 units for each proton removed,[29][30] as in the example of phosphoric acid above.
It can be seen in the table above that the second proton is removed from a negatively charged species. Since the proton carries a positive charge extra work is needed to remove it, which is why pKa2 is greater than pKa1. pKa3 is greater than pKa2 because there is further charge separation. When an exception to Pauling's rule is found, it indicates that a major change in structure is also occurring. In the case of VO+2(aq), the vanadium is octahedral, 6-coordinate, whereas vanadic acid is tetrahedral, 4-coordinate. This means that four "particles" are released with the first dissociation, but only two "particles" are released with the other dissociations, resulting in a much greater entropy contribution to the standard Gibbs free energy change for the first reaction than for the others.
Equilibrium pKa
![{\displaystyle {\ce {[VO2(H2O)4]+ <=> H3VO4 + H+ + 2H2O}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0b5fcdc28e4fbd98292ebd608643c231844a334f)







Isoelectric point
[edit]For substances in solution, the isoelectric point (pI) is defined as the pH at which the sum, weighted by charge value, of concentrations of positively charged species is equal to the weighted sum of concentrations of negatively charged species. In the case that there is one species of each type, the isoelectric point can be obtained directly from the pK values. Take the example of glycine, defined as AH. There are two dissociation equilibria to consider.
![{\displaystyle {\ce {AH2+<=>AH~+H+\qquad [AH][H+]={\mathit {K}}_{1}[AH2+]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/359f1d34ddc5ac4b4cecf45e11539bc14462e98f)
![{\displaystyle {\ce {AH<=>A^{-}~+H+\qquad [A^{-}][H+]={\mathit {K}}_{2}[AH]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/95c1af1bef047675675659473e3886aefff9fa79)
Substitute the expression for [AH] from the second equation into the first equation
![{\displaystyle {\ce {[A^{-}][H+]^{2}={\mathit {K}}_{1}{\mathit {K}}_{2}[AH2+]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/473772e02e925c83975026ce31b43a0a2dc4b1cc)
At the isoelectric point the concentration of the positively charged species, AH+2, is equal to the concentration of the negatively charged species, A−, so
![{\displaystyle [{\ce {H+}}]^{2}=K_{1}K_{2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3dfa9985f2d6769f35f98f98153ac6cabd9c011b)
Therefore, taking cologarithms, the pH is given by

pI values for amino acids are listed at proteinogenic amino acid. When more than two charged species are in equilibrium with each other a full speciation calculation may be needed.
Bases and basicity
[edit]The equilibrium constant Kb for a base is usually defined as the association constant for protonation of the base, B, to form the conjugate acid, HB+.

Using similar reasoning to that used before
![{\displaystyle {\begin{aligned}K_{\text{b}}&=\mathrm {\frac {[HB^{+}][OH^{-}]}{[B]}} \\\mathrm {p} K_{\text{b}}&=-\log _{10}\left(K_{\text{b}}\right)\end{aligned}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5dea1aac629a595476e18c042a8f4365a50f0efc)
Kb is related to Ka for the conjugate acid. In water, the concentration of the hydroxide ion, [OH−], is related to the concentration of the hydrogen ion by Kw = [H+][OH−], therefore
![{\displaystyle \mathrm {[OH^{-}]} ={\frac {K_{\mathrm {w} }}{\mathrm {[H^{+}]} }}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ab7f583da9f8b50145990ffa4342919930edfa16)
Substitution of the expression for [OH−] into the expression for Kb gives
![{\displaystyle K_{\text{b}}={\frac {[\mathrm {HB^{+}} ]K_{\text{w}}}{\mathrm {[B][H^{+}]} }}={\frac {K_{\text{w}}}{K_{\text{a}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/921c3abd37a1c5c00c31831509d3b090394c0d47)
When Ka, Kb and Kw are determined under the same conditions of temperature and ionic strength, it follows, taking cologarithms, that pKb = pKw − pKa. In aqueous solutions at 25 °C, pKw is 13.9965,[31] so

with sufficient accuracy for most practical purposes. In effect there is no need to define pKb separately from pKa,[32] but it is done here as often only pKb values can be found in the older literature.
For an hydrolyzed metal ion, Kb can also be defined as a stepwise dissociation constant

![{\displaystyle K_{\mathrm {b} }={\frac {[\mathrm {M} _{p}({\ce {OH}})_{q-1}^{+}][{\ce {OH-}}]}{[\mathrm {M} _{p}({\ce {OH}})_{q}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8391968e700e57f56f6041dd839f0fdb5e93780e)
This is the reciprocal of an association constant for formation of the complex.
Basicity expressed as dissociation constant of conjugate acid
[edit]Because the relationship pKb = pKw − pKa holds only in aqueous solutions (though analogous relationships apply for other amphoteric solvents), subdisciplines of chemistry like organic chemistry that usually deal with nonaqueous solutions generally do not use pKb as a measure of basicity. Instead, the pKa of the conjugate acid, denoted by pKaH, is quoted when basicity needs to be quantified. For base B and its conjugate acid BH+ in equilibrium, this is defined as
![{\displaystyle \mathrm {p} K_{\mathrm {aH} }(\mathrm {B} )=\mathrm {p} K_{\mathrm {a} }({\ce {BH+}})=-\log _{10}{\Big (}{\frac {[{\ce {B}}][{\ce {H+}}]}{[{\ce {BH+}}]}}{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b76149bc3dbc3d0375d6355bdf2342394a568776)
A higher value for pKaH corresponds to a stronger base. For example, the values pKaH (C5H5N) = 5.25 and pKaH ((CH3CH2)3N) = 10.75 indicate that (CH3CH2)3N (triethylamine) is a stronger base than C5H5N (pyridine).
Amphoteric substances
[edit]An amphoteric substance is one that can act as an acid or as a base, depending on pH. Water (below) is amphoteric. Another example of an amphoteric molecule is the bicarbonate ion HCO−3 that is the conjugate base of the carbonic acid molecule H2CO3 in the equilibrium

but also the conjugate acid of the carbonate ion CO2−3 in (the reverse of) the equilibrium

Carbonic acid equilibria are important for acid–base homeostasis in the human body.
An amino acid is also amphoteric with the added complication that the neutral molecule is subject to an internal acid–base equilibrium in which the basic amino group attracts and binds the proton from the acidic carboxyl group, forming a zwitterion.

At pH less than about 5 both the carboxylate group and the amino group are protonated. As pH increases the acid dissociates according to

At high pH a second dissociation may take place.

Thus the amino acid molecule is amphoteric because it may either be protonated or deprotonated.
Water self-ionization
[edit]The water molecule may either gain or lose a proton. It is said to be amphiprotic. The ionization equilibrium can be written

где в водном растворе H+ denotes a solvated proton. Often this is written as the hydronium ion H3O+, but this formula is not exact because in fact there is solvation by more than one water molecule and species such as H 5 O + 2 , H 7 O + 3 , и H 9 O + 4 также присутствуют. [ 33 ]
Константа равновесия дается
![{\ displayStyle K _ {\ text {a}} = \ mathrm {\ frac {[h^{+}] [OH^{-}]} {[h_ {2} o]}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/cdc540ad193c8f1661c1897698be93153fc5fb84)
С решениями, в которых концентрации растворенного вещества не очень высоки, концентрация [H 2 o] можно предположить, что это постоянное, независимо от растворенного вещества; Это выражение может быть заменено на
![{\ Dislaystyle k _ {{h}} = [\ mathrm}] [\ mathrm {OH} ^ {-}] \,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c0039f77db244ea2f6d03d3475dc7a232a8ccb16)
Таким образом, константа самоонизации воды, K W , является лишь особым случаем константы диссоциации кислоты. Логарифмическая форма, аналогичная P k a, также может быть определена

| T (° C) | 0 | 5 | 10 | 15 | 20 | 25 | 30 | 35 | 40 | 45 | 50 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P K W. | 14.943 | 14.734 | 14.535 | 14.346 | 14.167 | 13.997 | 13.830 | 13.680 | 13.535 | 13.396 | 13.262 |
Эти данные могут быть смоделированы параболой с

Из этого уравнения P K W = 14 при 24,87 ° C. При этой температуре и ионы водорода и гидроксида имеют концентрацию 10 −7 М.
Кислотность в невостовых решениях
[ редактировать ]Растворитель будет с большей вероятностью способствовать ионизации растворенной кислой молекулы при следующих обстоятельствах: [ 35 ]
- Это протетический растворитель , способный образовывать водородные связи.
- У него высокий номер донора , что делает его сильной базой Льюиса .
- Он имеет высокую диэлектрическую постоянную (относительную диэлектрическую проницаемость), что делает его хорошим растворителем для ионных видов.
P k Значения органических соединений часто получают с использованием апротонных растворителей диметилсульфоксид (ДМСО) [ 35 ] и ацетонитрил (ACN). [ 36 ]
| Растворитель | Номер донора [ 35 ] | Диэлектрическая постоянная [ 35 ] |
|---|---|---|
| Ацетонитрил | 14 | 37 |
| Диметилсульфоксид | 30 | 47 |
| Вода | 18 | 78 |
растворяет неполяриные гидрофобные ДМСО широко используется в качестве альтернативы воде, потому что он имеет более низкую диэлектрическую постоянную, чем воду, и является менее полярным и, таким образом, легче вещества. Он имеет измеримый p k диапазон от 1 до 30. Ацетонитрил менее прост, чем ДМСО, и, в целом, кислоты слабее, а основания в этом растворителе более сильнее. Некоторые значения P при a A 25 ° C для ацетонитрила (ACN) [ 37 ] [ 38 ] [ 39 ] и диметилсульфоксид (ДМСО). [ 40 ] показаны в следующих таблицах. Значения для воды включены для сравнения.
| Если ⇌ a − + H + | Акн | ДМСО | Вода |
|---|---|---|---|
| P -toluensulfonic | 8.5 | 0.9 | Сильный |
| 2,4-динитрофенол | 16.66 | 5.1 | 3.9 |
| Бензойная кислота | 21.51 | 11.1 | 4.2 |
| Уксусная кислота | 23.51 | 12.6 | 4.756 |
| Фенол | 29.14 | 18.0 | 9.99 |
| BH + ⇌ B + H + | Акн | ДМСО | Вода |
| Пирролидин | 19.56 | 10.8 | 11.4 |
| Триэтиламин | 18.82 | 9.0 | 10.72 |
| Протонная губка | 18.62 | 7.5 | 12.1 |
| Пиридин | 12.53 | 3.4 | 5.2 |
| Анилин | 10.62 | 3.6 | 4.6 |
Ионизация кислот меньше в кислотном растворителе, чем в воде. Например, хлорид водорода является слабой кислотой при растворенной уксусной кислоте . Это потому, что уксусная кислота является гораздо более слабым основанием, чем вода.

Сравните эту реакцию с тем, что происходит, когда уксусная кислота растворяется в более кислой растворителе чистой серной кислоты: [ 41 ]


Маловероятный геминальных диол виды CH 3 C (OH) + 2 является стабильным в этих средах. Для водных растворов масштаб рН является наиболее удобной функцией кислотности . [ 42 ] Другие функции кислотности были предложены для не вводных средств, наиболее заметной является функция кислотности Hammett , H 0 , для суперкидной среды и ее модифицированной версии h - для супербазных сред. [ 43 ]
В апротонных растворителях олигомеры , такие как хорошо известный димер уксусной кислоты , могут образовываться путем водородной связи. Кислота также может образовывать водородные связи с его сопряженным основанием. Этот процесс, известный как гомоконъюгация , имеет эффект усиления кислотности кислот, снижения их эффективных значений p a , путем стабилизации сопряженного основания. Гомоконъюгация усиливает протоно-доновую силу толуенсульфоновой кислоты в растворе ацетонитрила почти 800. [ 44 ]
В водных растворах гомоконъюгация не происходит, потому что вода образует более сильные водородные связи с конъюгатным основанием, чем кислота.
Смешанные растворители
[ редактировать ]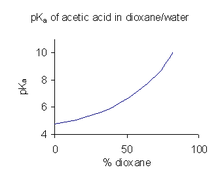
Когда соединение имеет ограниченную растворимость в воде, это обычная практика (например, в фармацевтической промышленности) для определения значений P в a смеси растворителя, таких как вода/ диоксан или вода/ метанол , в которых соединение является более растворимым. [ 46 ] В примере, показанном справа, значение p a a Значение круто возрастает с увеличением процента диоксана, поскольку диэлектрическая проницаемость смеси уменьшается.
Значение P K Значение , полученное в смешанном растворителе, не может быть использовано непосредственно для водных растворов. Причина этого заключается в том, что когда растворитель находится в его стандартном состоянии, его активность определяется как единица. Например, стандартное состояние воды: диоксановая смесь с коэффициентом смешивания 9: 1 именно такая смесь растворителя без добавленных растворенных веществ. P k Чтобы получить значение для использования с водными растворами, он должен быть экстраполирован до нулевой концентрации совластного растворителя из значений, полученных из различных сочетания смесей.
Эти факты скрыты из -за упущения растворителя от выражения, которое обычно используется для определения P k a , но значения P a a a значения, полученные в данном смешанном растворителе, можно сравнить друг с другом, придавая относительную силу кислоты. То же самое относится и к значениям P a , полученные в конкретном неводном растворителе, таких как ДМСО.
Универсальный, не независимый от растворителя масштаб для константы диссоциации кислоты не был разработан, поскольку не существует известного способа сравнения стандартных состояний двух различных растворителей.
Факторы, которые влияют на a p значения
[ редактировать ]Второе правило Полинга состоит в том, что значение первого p k a для кислот формулы XO m (OH) N зависит главным образом от количества групп Oxo M , и приблизительно независимо от количества гидрокси групп N , а также от Центральный атом X. Приблизительные значения P k a составляют 8 для m = 0, 2 для m = 1, −3 для m = 2 и <-10 для m = 3. [ 28 ] В качестве альтернативы были предложены различные численные формулы, включая P k a = 8 - 5 м (известный как правило Белла ), [ 29 ] [ 47 ] p k a = 7 - 5 м , [ 30 ] [ 48 ] или p k a = 9 - 7 м . [ 29 ] Зависимость от М коррелирует с состоянием окисления центрального атома, x: чем выше состояние окисления, тем сильнее оксикид.
Например, p k a для Hclo составляет 7,2, для Hclo 2 составляет 2,0, для Hclo 3 - -1, а Hclo 4 - сильная кислота ( P k a ≪ 0 ). [ 7 ] Повышенная кислотность при добавлении группы OXO обусловлена стабилизацией конъюгатного основания путем делокализации его негативного заряда в течение дополнительного атома кислорода. [ 47 ] Это правило может помочь назначить молекулярную структуру: например, фосфорная кислота , имеющая молекулярную формулу H 3 PO 3 , имеет AP K A Rese 2, что предполагало, что структура составляет HPO (OH) 2 , как позже подтверждено спектроскопией ЯМР , а не P (OH) 3 , который, как ожидается, будет иметь AP K A около 8. [ 48 ]

Индуктивные эффекты и мезомерные эффекты влияют на значения p a . Простой пример предоставляется влиянием замены атомов водорода в уксусной кислоте более электроотрицательным атомом хлора. Электронный эффект заместителя облегчает ионизацию, поэтому последовательные значения p уменьшаются a в серии 4.7, 2,8, 1,4 и 0,7, когда присутствует 0, 1, 2 или 3 атома хлора. [ 49 ] Уравнение Хамметт обеспечивает общее выражение для эффекта заместителей. [ 50 ]
- log ( k a ) = log ( k 0
а ) + р.
K a - константа диссоциации замещенного соединения, k 0
A является постоянной диссоциации, когда заместитель является водородом, ρ является свойством необоснащенного соединения, а σ имеет определенное значение для каждого заместителя. График журнала ( k a ) против σ - прямая линия с журналом перехвата ( k 0
а ) и наклон ρ. Это пример линейного отношения свободной энергии , поскольку log ( k a ) пропорциональна стандартному изменению свободной энергии. Хамметт изначально [ 51 ] взаимосвязь с данными кислоты с различными заместителями в орто -пара -позициях бензойной Сформулировал : некоторые числовые значения находятся в уравнении Хамметт . Это и другие исследования позволили заказать заместители в соответствии с их электронно-гигинной или электронно-выделяющей силой, а также различать индуктивные и мезомерные эффекты. [ 52 ] [ 53 ]
Обычно спирты не ведут себя как кислоты в воде, но наличие двойной связи, прилегающей к группе ОН, может существенно снизить P a a с помощью механизма таутомеризма кето -энола . Аскорбиновая кислота является примером этого эффекта. Дикетон 2,4-пентанедион ( ацетилацетон ) также является слабой кислотой из-за равновесия кето-эенол. В ароматических соединениях, таких как фенол , которые имеют заместитель ОН, сопряжение с ароматическим кольцом в целом значительно увеличивает стабильность депротонированной формы.

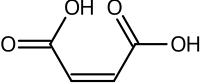
Структурные эффекты также могут быть важными. Разница между фумариновой кислотой и малеиновой кислотой является классическим примером. Фумариновая кислота составляет (E) -1,4-but-2-enedioic кислоты, транс -изомер , тогда как малеиновая кислота является соответствующим цис- , т.е. изомером транс изомерия ). Фумариновая кислота имеет значения P a a приблизительно 3,0 и 4,5. Напротив, малеиновая кислота имеет значения P a a приблизительно 1,5 и 6,5. Причиной такого большого различия является то, что когда один протон удаляется из цис -изомер (малеиновая кислота) сильная внутримолекулярная водородная связь с близлежащей оставшейся карбоксильной группой. Это способствует формированию малеата h + , и это выступает против удаления второго протона из этого вида. В транс -изомере две карбоксильные группы всегда далеко друг от друга, поэтому водородная связь не наблюдается. [ 54 ]
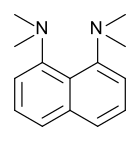
Proton Sponge , 1,8-бис (диметиламино) нафталин, имеет ap k значение 12,1. Это одна из самых сильных известных амина. Высокая основность объясняется облегчением деформации при протонации и сильной внутренней водородной связи. [ 55 ] [ 56 ]
В этом разделе следует также упомянуть эффекты растворителя и сольватации. Оказывается, эти влияния более тонкие, чем у диэлектрической среды, упомянутой выше. Например, ожидаемое (электронные эффекты метильных заместителей) и наблюдается в газовой фазах базовой основы метиламина, ME 3 N> ME 2 NH> MenH 2 > NH 3 , изменяется водой для меня 2 NH> Menh 2 > Я 3 н> нх 3 . Нейтральные молекулы метиламина связаны с водородом с молекулами воды в основном через один акцептор, N-HOH, взаимодействие и лишь изредка только одну еще одну донорскую связь, NH-OH 2 . Следовательно, метиламины стабилизируются примерно в той же степени путем гидратации, независимо от количества метильных групп. В отличие от этого, соответствующие катионы метиламмония всегда используют все доступные протоны для связи с донором NH -OH 2 . Относительная стабилизация ионов метиламмония, таким образом, уменьшается с количеством метильных групп, объясняющих порядок базовой воды метиламинов. [ 4 ]
Термодинамика
[ редактировать ]Константа равновесия связана со стандартным изменением энергии Гиббса для реакции, поэтому для константы диссоциации кислоты
- .

R - это постоянная газа , а T - абсолютная температура . Обратите внимание, что p k a = −log ( k a ) и 2,303 ≈ ln (10) . При 25 ° C, Δ G ⊖ в KJ · моль −1 ≈ 5,708 p k a (1 кДж · моль −1 = 1000 джоулей на моль ). Свободная энергия состоит из энтальпийного термина и энтропийного термина. [ 11 ]

Стандартное изменение энтальпии может быть определена с помощью калориметрии или с помощью уравнения Van 'T HOFF , хотя калориметрический метод предпочтительнее. При определении как стандартного изменения энтальпии, так и константы диссоциации кислоты, стандартное изменение энтропии легко рассчитывается из приведенного выше уравнения. следующей таблице термины энтропии рассчитываются по экспериментальным значениям p a В и Δ h ⊖ Полем Данные были выбраны критически и обращаются к 25 ° C и нулевой ионной прочности в воде. [ 11 ]
| Сложный | Равновесие | P K A. | Δ G. ⊖ (KJ · раз −1 ) [ D ] | Δ H. ⊖ (KJ · раз −1 ) | - T Δ S ⊖ (KJ · раз −1 ) [ E ] |
|---|---|---|---|---|---|
| Ха = уксусная кислота | Ха ⇌ ч + + А − | 4.756 | 27.147 | −0.41 | 27.56 |
| H 2 а + = Глицин h + | H 2 а + ⇌ Ha + h + | 2.351 | 13.420 | 4.00 | 9.419 |
| Ха ⇌ ч + + А − | 9.78 | 55.825 | 44.20 | 11.6 | |
| H 2 A = Малеиновая кислота | H 2 a ⇌ ха − + H + | 1.92 | 10.76 | 1.10 | 9.85 |
| Ха − ⇌ h + + А 2− | 6.27 | 35.79 | −3.60 | 39.4 | |
| H 3 A = лимонная кислота | H 3 A ⇌ H 2 A − + H + | 3.128 | 17.855 | 4.07 | 13.78 |
| H 2 а − ⇌ ха 2− + H + | 4.76 | 27.176 | 2.23 | 24.9 | |
| Ха 2− ⇌ a 3− + H + | 6.40 | 36.509 | −3.38 | 39.9 | |
| H 3 A = борная кислота | H 3 A ⇌ H 2 A − + H + | 9.237 | 52.725 | 13.80 | 38.92 |
| H 3 A = фосфорная кислота | H 3 A ⇌ H 2 A − + H + | 2.148 | 12.261 | −8.00 | 20.26 |
| H 2 а − ⇌ ха 2− + H + | 7.20 | 41.087 | 3.60 | 37.5 | |
| Ха 2− ⇌ a 3− + H + | 12.35 | 80.49 | 16.00 | 54.49 | |
| Ха − = Водород серо | Ха − ⇌ a 2− + H + | 1.99 | 11.36 | −22.40 | 33.74 |
| H 2 A = щавелевая кислота | H 2 a ⇌ ха − + H + | 1.27 | 7.27 | −3.90 | 11.15 |
| Ха − ⇌ a 2− + H + | 4.266 | 24.351 | −7.00 | 31.35 |
- ^ Ион водорода не существует как таковой в растворе. Он в сочетании с молекулой растворителя; Когда растворитель - это вода, гидрония : образуется ион ЧАС + + H 2 O → H 3 O + Полем Эта реакция является количественной и, следовательно, может быть проигнорирована в контексте химического равновесия.
- ^ Обычная практика - цитировать значения p k, а не значения k . p k = -log 10 k . P K A часто называют константой диссоциации кислоты, но это, строго говоря, неправильно, поскольку P K A является кологарифмом постоянной диссоциации.
- ^ В этом определении подразумевается, что отношение коэффициентов активности , является постоянной со значением 1 в данном наборе экспериментальных условий.
- ^ Δ G. ⊖ ≈ 2,303 RT P K A
- ^ Вычисленные здесь, из значений Δ и Δ g, поставляемых в цитировании, с использованием - t Δ s ⊖ = Δ g ⊖ - Δ H. ⊖

| Сложный | Равновесие | P K A. | ΔH ⊖ (KJ · раз −1 ) | - T Δ S ⊖ (KJ · раз −1 ) |
|---|---|---|---|---|
| B = аммиак | HB + ⇌ B + H + | 9.245 | 51.95 | 0.8205 |
| B = метиламин | HB + ⇌ B + H + | 10.645 | 55.34 | 5.422 |
| B = триэтиламин | HB + ⇌ B + H + | 10.72 | 43.13 | 18.06 |
Первая точка, которую следует отметить, заключается в том, что, когда P k a является положительным, стандартное изменение свободной энергии для реакции диссоциации также является положительным. Во -вторых, некоторые реакции являются экзотермическими , а некоторые являются эндотермическими , но, когда Δ H ⊖ отрицательный t ΔS ⊖ является доминирующим фактором, который определяет, что Δ G ⊖ положительный. Наконец, вклад энтропии всегда неблагоприятный ( Δ s ⊖ <0 ) в этих реакциях. Ионы в водном растворе имеют тенденцию ориентировать окружающие молекулы воды, которые заказывают раствор и уменьшают энтропию. Вклад иона в энтропию - это частичная молярная энтропия, которая часто является отрицательной, особенно для небольших или высоко заряженных ионов. [ 57 ] Ионизация нейтральной кислоты включает образование двух ионов, чтобы энтропия уменьшалась ( Δ s ⊖ <0 ). На второй ионизации той же кислоты сейчас есть три иона, а анион имеет заряд, поэтому энтропия снова уменьшается.
Обратите внимание, что стандартное изменение свободной энергии для реакции заключается в изменениях от реагентов в их стандартных состояниях до продуктов в их стандартных состояниях. Изменение свободной энергии в равновесии равен нулю, поскольку химические потенциалы реагентов и продуктов равны в равновесии.
Экспериментальное определение
[ редактировать ]
Экспериментальное определение значений p a обычно выполняется с помощью титрования , в среде высокой ионной прочности и при постоянной температуре. [ 58 ] Типичная процедура была бы следующей. Раствор соединения в среде подкисляется сильной кислотой до точки, где соединение полностью протонируется. Затем решение титруют сильным основанием до тех пор, пока все протоны не будут удалены. В каждой точке в титровании рН измеряется с использованием стеклянного электрода и pH -метра . Константы равновесия обнаруживаются путем подгонки расчетных значений рН для наблюдаемых значений, используя метод наименьших квадратов . [ 59 ]
Общий объем добавленной сильной основы должен быть небольшим по сравнению с начальным объемом раствора титрана, чтобы сохранить ионную прочность почти постоянной. Это гарантирует, что P K A остается инвариантным во время титрования.
Рассчитанная кривая титрования для щавелевой кислоты показана справа. Оксальновая кислота имеет значения P 1,27 a и 4,27. Следовательно, буферные районы будут центрироваться при рН 1,3 и рН 4,3. Регионы буфера несут информацию, необходимую для получения значений P a , в качестве концентрации кислоты и конъюгатной базы изменяются вдоль буферной области.
Между двумя буферными областями существует конечная точка или точка эквивалентности , примерно при рН 3. Эта конечная точка не является резкой и типична для дипротечной кислоты, чьи буферные области перекрываются небольшим количеством: P k a2 -p k k A1 в этом примере составляет около трех. (Если бы разница в значениях P k была примерно на два или менее, конечная точка не была бы заметной.) Вторая конечная точка начинается с рН 6,3 и является резкой. Это указывает на то, что все протоны были удалены. Когда это так, решение не буферируется, а pH круто поднимается при добавлении небольшого количества сильного основания. Тем не менее, pH не продолжает расти на неопределенный срок. Новая буферная область начинается примерно с pH 11 (p k w -3), где самоонизация воды становится важной.
Очень трудно измерить значения pH менее двух в водном растворе стеклом электродом , потому что уравнение Нернста разрушается при таких низких значениях pH. Для определения значений p k менее 2 или более 11 спектрофотометрических [ 60 ] [ 61 ] или ЯМР [ 62 ] [ 63 ] Измерения могут использоваться вместо измерений pH или в сочетании.
Когда стеклянный электрод не может быть использован, как и в случае неводных растворов, часто используются спектрофотометрические методы. [ 38 ] Они могут включать измерения поглощения или флуоресценции . В обоих случаях предполагается, что измеренная величина пропорциональна сумме вкладов от каждого фотоактивного вида; При измерениях поглощения предполагается, что закон пива -ламберта применяется.
Изотермическая титровая калориметрия (ITC) может использоваться для определения как AP -значения , так и соответствующей стандартной энтальпии для диссоциации кислоты. [ 64 ] Программное обеспечение для выполнения расчетов поставляется производителями приборов для простых систем.
Водные растворы с нормальной водой не могут быть использованы для 1 H ЯМР измерения, но тяжелая вода , D 2 O , вместо этого должен использоваться. 13 C ЯМР, однако, можно использовать с нормальной водой и 1 Спектры ЯМР можно использовать со средами не ведущими. Количество, измеренные с помощью ЯМР, являются усредненными по времени химических сдвигов , так как протонный обмен быстр в масштабе времени ЯМР. Другие химические сдвиги, такие как сдвиги 31 P может быть измерено.
Микроконтанта
[ редактировать ]
Для некоторых полипротичных кислот диссоциация (или ассоциация) происходит в более чем один неравственный сайт, [ 4 ] и наблюдаемая константа макроскопического равновесия, или макроконстанта, представляет собой комбинацию микроконстатировки с участием различных видов. Когда один реагент образует два продукта параллельно, макроконстатинг представляет собой сумму из двух микроконтанта, Это верно, например, для депротонирования аминокислотного цистеина , который существует в растворе в качестве нейтрального Zwitterion Hs -ch 2 −ch (nh + 3 ) −coo − Полем Два микроконстатирирования представляют депротонирование либо в серре, либо в азоте, а макросоистовая сумма здесь-константа с кислотой диссоциации [ 65 ]



Точно так же база, такая как спермин, имеет более одного сайта, где может происходить протонирование. Например, монопротонирование может происходить на терминале −nh 2 группа или во внутренней −nh- группы. Значения K B для диссоциации спермин-протонированных на одном или другом из участков являются примерами микроконтанта . Они не могут быть определены непосредственно с помощью рН, поглощения, флуоресценции или измерений ЯМР; Измеренное значение K B -это сумма значений k для микрореакций.

Тем не менее, сайт протонирования очень важен для биологической функции, поэтому были разработаны математические методы для определения микроконстатировки. [ 66 ]
Когда два реагента образуют один продукт параллельно, макроконстати [ 65 ] Например, вышеупомянутое равновесие для спермина может рассматриваться в терминах значений k A двух таутомерных конъюгатных кислот, с макроконстатонами в этом случае Это эквивалентно предыдущему выражению, так как пропорционально




Когда реагент подвергается двум реакциям последовательно, макро-контейнер для комбинированной реакции является произведением микроконстатирования для двух этапов. Например, вышеупомянутое цистеиновое Zwitterion может потерять два протона, один из серы и один из азота, а общий макросодержащий для потери двух протонов-продукт двух константов диссоциации [ 65 ] Это также может быть написано с точки зрения логарифмической константы как


Приложения и значение
[ редактировать ]Знание значений p a a важно для количественной обработки систем, включающих кислотные базовые равновесия в растворе. Многие заявки существуют в биохимии ; Например, значения P a a белков и аминокислотных боковых цепей имеют большое значение для активности ферментов и стабильности белков. [ 67 ] белка P k a Значения не всегда могут быть измерены напрямую, но могут быть рассчитаны с использованием теоретических методов. Буферные растворы широко используются для обеспечения растворов при физиологическом pH или вблизи изучения биохимических реакций; [ 68 ] Дизайн этих решений зависит от знания значений p a a a значения их компонентов. Важные буферные растворы включают MOP , который обеспечивает раствор с рН 7,2 и трицина , который используется в гелевом электрофорезе . [ 69 ] [ 70 ] Буферизация является неотъемлемой частью физиологии кислотного основания, включая кислотный гомеостаз , [ 71 ] и является ключом к пониманию расстройств, таких как кислотное расстройство . [ 72 ] [ 73 ] [ 74 ] Изоэлектрическая точка данной молекулы является функцией его значений P K , поэтому разные молекулы имеют разные изоэлектрические точки. Это позволяет методику, называемую изоэлектрической фокусировкой , [ 75 ] который используется для разделения белков с помощью двухмерного гель-полиакриламидного геля электрофореза .
Буферные решения также играют ключевую роль в аналитической химии . Они используются всякий раз, когда необходимо исправить рН решения в определенном значении. По сравнению с водным раствором рН буферного раствора относительно нечувствителен к добавлению небольшого количества сильного кислотного или сильного основания. Буферная емкость [ 76 ] простого буферного раствора наибольшее, когда pH = P k a . При экстракции кислотной базы эффективность экстракции соединения в органическую фазу, такую как эфир , может быть оптимизирована путем регулировки pH водной фазы с помощью соответствующего буфера. При оптимальном рН концентрация электрически нейтрального вида максимизируется; Такой вид более растворим в органических растворителях, имеющих низкую диэлектрическую постоянную, чем в воде. Этот метод используется для очистки слабых кислот и оснований. [ 77 ]
Индикатор pH - это слабая кислотная или слабая основание, которое изменяет цвет в диапазоне перехода pH, который составляет приблизительно p k a ± 1. Конструкция универсального индикатора требует смесь индикаторов, p значения смежные так что их переходные рН просто перекрываются.
В фармакологии ионизация соединения изменяет его физическое поведение и свойства макроса, такие как растворимость и липофильность , log p ). Например, ионизация любого соединения увеличит растворимость в воде, но уменьшит липофильность. Это используется в разработке лекарств, чтобы увеличить концентрацию соединения в крови путем корректировки P K A ионизируемой группы. [ 78 ]
Знание p k a значения важно для понимания координационных комплексов , которые формируются в результате взаимодействия иона металла, m M+ , действуя как кислота Льюиса , с лигандом , L, действуя в качестве основания Льюиса . Тем не менее, лиганд может также подвергаться протонационным реакциям, поэтому образование комплекса в водном растворе может быть символически представлено реакцией
![{\ Displaystyle [{\ ce {m (h2o) _ {\ mathit {n}}}}]^{m+}+{\ ce {lh <=>}} \ [{\ ce {m (h2o)}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}} _ {n-1} {\ ce {l}}]^{(m-1)+}+{\ ce {h3o+}}}}}}}}}}}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/696ff04eb241a2f6a358b8dd1b9c373ea3a8c91d)
Чтобы определить константу равновесия для этой реакции, в которой лиганд теряет протон, должен быть известен P K A протонированного лиганда. На практике лиганд может быть полипротичным; Например, ЭДТА 4− может принять четыре протона; В этом случае все значения P должны K быть известны. Кроме того, ион металлов подвержен гидролизу , то есть он ведет себя как слабая кислота, поэтому также должны быть известны значения P k для реакций гидролиза. [ 79 ]
Оценка опасности, связанной с кислотой или базой, может потребовать знания значений p a . [ 80 ] Например, цианид водорода является очень токсичным газом, потому что ион цианида ингибирует железосодержащий фермент цитохром С оксидазу . Кианид водорода является слабой кислотой в водном растворе с AP K A около 9. В сильно щелочных растворах выше рН 11, скажем, следует, что цианид натрия «полностью диссоциируется», поэтому опасность из -за цианистового газа водорода сильно снижена Полем Кислотный раствор, с другой стороны, очень опасен, потому что весь цианид находится в его кислотной форме. Прием цианида во рту потенциально летально, независимо от рН, из -за реакции с цитохромом С оксидазой.
В экологических науках кислотные равновесия важны для озер [ 81 ] и реки; [ 82 ] [ 83 ] Например, гуминовые кислоты являются важными компонентами природных вод. Другой пример происходит в химической океанографии : [ 84 ] Чтобы количественно оценить растворимость железа (III) в морской воде при различных соленях , значения P K A для образования гидролиза железо (III) Fe (OH) 2+ , Fe (OH) + 2 и Fe (OH) 3 были определены вместе с продуктом растворимости гидроксида железа . [ 85 ]
Значения для общих веществ
[ редактировать ]Существует несколько методов для определения P a a химического вещества, что приводит к некоторым несоответствиям между различными источниками. Хорошо измеренные значения обычно находятся в пределах 0,1 единиц друг от друга. Данные, представленные здесь, были взяты при 25 ° С в воде. [ 7 ] [ 86 ] Больше значений можно найти в разделе термодинамики выше. Таблица P K A углеродных кислот, измеренная в ДМСО, может быть найдена на странице на Карбанионах .
| Химический | Равновесие | P K A. |
|---|---|---|
| BH = аденин | BH + 2 ⇌ b + h + |
4.17 |
| BH ⇌ б - + H + | 9.65 | |
| H 3 A = арсениновая кислота | H 3 A ⇌ H 2 A − + H + | 2.22 |
| H 2 а − ⇌ ха 2− + H + | 6.98 | |
| Ха 2− ⇌ a 3− + H + | 11.53 | |
| Ха = бензоевая кислота | Ха ⇌ ч + + А − | 4.204 |
| HA = батарикальная кислота | Ха ⇌ ч + + А − | 4.82 |
| H 2 A = хромовая кислота | H 2 a ⇌ ха − + H + | 0.98 |
| Ха − ⇌ a 2− + H + | 6.5 | |
| B = кодеин | BH + ⇌ B + H + | 8.17 |
| Ха = Крезол | Ха ⇌ ч + + А − | 10.29 |
| Ха = муравьиная кислота | Ха ⇌ ч + + А − | 3.751 |
| HA = гидрофторическая кислота | Ха ⇌ ч + + А − | 3.17 |
| HA = гидроциановая кислота | Ха ⇌ ч + + А − | 9.21 |
| HA = водород Селенид | Ха ⇌ ч + + А − | 3.89 |
| HA = перекись водорода (90%) | Ха ⇌ ч + + А − | 11.7 |
| Ха = молочная кислота | Ха ⇌ ч + + А − | 3.86 |
| Ха = пропионовая кислота | Ха ⇌ ч + + А − | 4.87 |
| Ха = фенол | Ха ⇌ ч + + А − | 9.99 |
| H 2 A = L -(+) -аскорбиновая кислота | H 2 a ⇌ ха − + H + | 4.17 |
| Ха − ⇌ a 2− + H + | 11.57 |
Смотрите также
[ редактировать ]- Ацидоз
- Кислоты в вине : тартарическая , яростная и лимонка являются основными кислотами в вине.
- Алкалоз
- Артериальная крови газ
- Химическое равновесие
- Проводимость (электролитическая)
- Механизм Grotthuss : как протоны передаются между ионами гидрония и молекулами воды, учитывая исключительно высокую ионную подвижность протона (анимация).
- Функция кислотности Хамметт : мера кислотности, которая используется для очень концентрированных растворов сильной кислоты, включая супецид .
- Ионовый транспортный номер
- Океанский подкисление : растворение атмосферного углекислого газа влияет на PH морской воды . Реакция зависит от общего неорганического углерода и от растворимости равновесия с твердыми карбонатами, такими как известняк и доломит .
- Закон разведения
- PCO2
- pH
- Диаграмма преобладания : относится к равновесию с участием полиоксейнионов . P k Значения необходимы для построения этих диаграмм.
- Аффинность протонов : мера основной в газовой фазе.
- Константы стабильности комплексов : образование комплекса часто можно рассматривать как конкуренция между ионом протона и металла для лиганда, который является продуктом диссоциации кислоты.
Примечания
[ редактировать ]Ссылки
[ редактировать ]- ^ Уиттен, Кеннет У.; Гейли, Кеннет Д.; Дэвис, Рэймонд Э. (1992). Общая химия (4 -е изд.). Saunders College Publishing. п. 660 . ISBN 0-03-072373-6 .
- ^ Petrucci, Ralph H.; Харвуд, Уильям С.; Херринг, Ф. Джеффри (2002). Общая химия (8 -е изд.). Прентис Холл. С. 667–8 . ISBN 0-13-014329-4 .
- ^ Perrin DD, Dempsey B, Serjeant EP (1981). «Глава 3: Методы P K прогнозирование ». P K прогноз для органических кислот и оснований . (вторичный). Лондон: Чепмен и Холл. С. 21–26. doi : 10.1007/978-94-009-5883-8 . ISBN 978-0-412-22190-3 .
- ^ Jump up to: а беременный в Fraczkiewicz R (2013). «При прогнозировании ионизации кремнезема». В Reedijk J (ред.). Спортивный модуль в химии, молекулярных науках и химической технике . (вторичный). Справочный модуль в химии, молекулярных науках и химической инженерии [онлайн] . Тол. 5. Амстердам, Нидерланды: Elsevier. doi : 10.1016/b978-0-12-409547-2.02610-x . ISBN 9780124095472 .
- ^ Miessler, Gary L.; Тарр, Дональд А. (1991). Неорганическая химия (2 -е изд.). Прентис Холл. ISBN 0-13-465659-8 Полем Глава 6: Химия кислотной базы и донор -акцептор
- ^ Jump up to: а беременный Белл, RP (1973). Протон в химии (2 -е изд.). Лондон: Чепмен и Холл. ISBN 0-8014-0803-2 Полем Включает обсуждение многих органических кислот Бренстеда.
- ^ Jump up to: а беременный в Шрайвер, DF; Аткинс, PW (1999). Неорганическая химия (3 -е изд.). Оксфорд: издательство Оксфордского университета. ISBN 0-19-850331-8 Полем Глава 5: Кислоты и основания
- ^ Housecroft, CE; Sharpe, AG (2008). Неорганическая химия (3 -е изд.). Прентис Холл. ISBN 978-0-13-175553-6 Полем Глава 6: Кислоты, основания и ионы в водном растворе
- ^ Headrick, JM; Дикен, например; Уолтерс, RS; Молоток, ни; Кристи, Ра; Cui, J.; Мишакин, их; Дункан, Массачусетс; Джонсон, Массачусетс; Джордан, К.Д. (2005). «Спектральные сигнатуры гидратированных протонных вибраций в кластерах воды». Наука . 308 (5729): 1765–69. Bibcode : 2005sci ... 308.1765H . doi : 10.1126/science.1113094 . PMID 15961665 . S2CID 40852810 .
- ^ Smiechowski, M.; Stangret, J. (2006). «Гидратация протонов в водном растворе: инфракрасные исследования Фурье -преобразования спектров HDO». J. Chem. Физический 125 (20): 204508–204522. BIBCODE : 2006JCHPH.125T4508S . doi : 10.1063/1,2374891 . PMID 17144716 .
- ^ Jump up to: а беременный в Голдберг, Р.; Кишор, Н.; Леннен Р. (2002). «Термодинамические величины для ионизационных реакций буферов» (PDF) . J. Phys Химический Рефери Данные . 31 (2): 231–370. Bibcode : 2002jpcrd..31..231g . doi : 10.1063/1.1416902 . Архивировано из оригинала (PDF) на 2008-10-06.
- ^ Джолли, Уильям Л. (1984). Современная неорганическая химия . МакГроу-Хилл. С. 198 . ISBN 978-0-07-032760-3 .
- ^ Burgess, J. (1978). Ионы металлов в растворе . Эллис Хорвуд. ISBN 0-85312-027-7 Полем В разделе 9.1 «Кислотность сольватированных катионов» перечисляется много значений P a .
- ^ Petrucci, RH; Harwood, Rs; Херринг, Ф.Г. (2002). Общая химия (8 -е изд.). Прентис Холл. ISBN 0-13-014329-4 Полем с.698
- ^ Jump up to: а беременный Россотти, FJC; Россотти, Х. (1961). Определение констант стабильности . МакГроу - Хилл. Глава 2: Коэффициенты активности и концентрации, стр. 5-10
- ^ «Проект: ионная коррекция прочности для констант стабильности» . Международный союз чистой и прикладной химии . Получено 2019-03-28 .
- ^ Россотти, Фрэнсис Дж. С; Розотти, Хейзел (1961). Определение констант стабильности: и другие константы равновесия в растворе . Нью-Йорк: МакГроу-Хилл. С. 5–10. ISBN 9781013909146 Полем Архивировано из оригинала 7 февраля 2020 года.
- ^ Аткинс, PW; Де Паула, Дж. (2006). Физическая химия . Издательство Оксфордского университета. ISBN 0-19-870072-5 Полем Раздел 7.4: Ответ равновесия на температуру
- ^ Petrucci, Ralph H.; Харвуд, Уильям С.; Херринг, Ф. Джеффри (2002). Общая химия: принципы и современные применения (8 -е изд.). Прентис Холл. п. 633 . ISBN 0-13-014329-4 Полем
Вам интересно ... как использование действий делает равновесие постоянную бессмысленность?
- ^ Шрайвер, DF; Аткинс, PW (1999). Неорганическая химия (3 -е изд.). Издательство Оксфордского университета. ISBN 0-19-850331-8 Полем Раздел 5.1C Сильные и слабые кислоты и основания
- ^ Портерфилд, Уильям В. (1984). Неорганическая химия . Аддисон-Уэсли. п. 260. ISBN 0-201-05660-7 .
- ^ Jump up to: а беременный Шрайвер, DF; Аткинс, PW (1999). Неорганическая химия (3 -е изд.). Издательство Оксфордского университета. ISBN 0-19-850331-8 Полем Раздел 5.2 Выравнивание растворителя
- ^ Леванов, Ав; Исаикина, О. Я.; Lunin, VV (2017). «Константа диссоциации азотной кислоты». Российский журнал физической химии а . 91 (7): 1221–1228. BIBCODE : 2017RJPCA..91.1221L . doi : 10.1134/s0036024417070196 . S2CID 104093297 .
- ^ Trummal, Александр; Lipping, Lauri; Калджуранд, Ивари; Koppel, Ilmar A.; Лейто, Иво (2016). «Кислотность сильных кислот в воде и диметилсульфоксиде». Журнал физической химии а . 120 (20): 3663–3669. BIBCODE : 2016JPCA..120.3663T . doi : 10.1021/acs.jpca.6b02253 . PMID 27115918 . S2CID 29697201 .
- ^ Мехта, Акул (22 октября 2012 г.). «Уравнение Хендерсона -Хассельбалх: вывод P K A и P K B » . Pharmaxchange . Получено 16 ноября 2014 года .
- ^ Значения для 25 ° C и 0 ионной силы - Пауэлл, Киптон Дж.; Браун, Пол Л.; Бирн, Роберт Х.; Гайда, Тамас; Хефтер, Гленн; Шеберг, Стафан; Wanner, Hans (2005). "Химическое видообразование экологически значимых тяжелых металлов с неорганическими лигандами. Часть 1: HG 2+ - кл − , ОЙ − , Co 3 2- , Так 4 2- и PO 4 3- Водные системы » . Pure Appl. Chem . 77 (4): 739–800. DOI : 10.1351/pac200577040739 .
- ^ Браун, те; Лемей, он; Взрываться, быть; Мерфи, C.; Вудворд, П. (2008). Химия: центральная наука (11 -е изд.). Нью-Йорк: Прентис-Холл. п. 689. ISBN 978-0-13-600617-6 .
- ^ Jump up to: а беременный Гринвуд, NN; Эрншоу А. (1997). Химия элементов (2 -е изд.). Оксфорд: Баттерворт-Хейнеманн. п. 50. ISBN 0-7506-3365-4 .
- ^ Jump up to: а беременный в Miessler, Gary L.; Тарр Дональд А. (1999). Неорганическая химия (2 -е изд.). Прентис Холл. п. 164. ISBN 0-13-465659-8 .
- ^ Jump up to: а беременный Huheey, Джеймс Э. (1983). Неорганическая химия (3 -е изд.). Harper & Row. п. 297. ISBN 0-06-042987-9 .
- ^ Lide, DR (2004). Справочник по химии и физике CRC, Студенческое издание (84 -е изд.). CRC Press. ISBN 0-8493-0597-7 Полем Раздел D - 152
- ^ Skoog, Douglas A.; Запад, Дональд М.; Холлер, Ф. Джеймс; Крауч, Стэнли Р. (2014). Основы аналитической химии (9 -е изд.). Брукс/Коул. п. 212. ISBN 978-0-495-55828-6 .
- ^ Housecroft, CE; Sharpe, AG (2004). Неорганическая химия (2 -е изд.). Прентис Холл. п. 163. ISBN 978-0-13-039913-7 .
- ^ Harned, HS; Оуэн, BB (1958). Физическая химия электролитических растворов . Нью -Йорк: Reinhold Publishing Corp. с. 634 –649, 752–754.
- ^ Jump up to: а беременный в дюймовый Лоудон, Г. Марк (2005), Органическая химия (4 -е изд.), Нью -Йорк: издательство Оксфордского университета, с. 317–318, ISBN 0-19-511999-1
- ^ Март, J .; Смит, М. (2007). Усовершенствованная органическая химия (6 -е изд.). Нью -Йорк: Джон Уайли и сыновья. ISBN 978-0-471-72091-1 Полем Глава 8: Кислоты и основания
- ^ Kütt, A.; Movchun, v.; Родима, т; Dansauer, T.; Русанов, EB; Лейто, я.; Калджуранд, я.; Koppel, J.; Pihl, V.; Koppel, я.; Ovsjannikov, G.; TOOM, L.; Мишима, м.; Medebielle, M.; Лорк, E.; Röschenthaler, Gv.; Коппель, ИА; Kolomeitsev, AA (2008). «Пентакис (трифторметил) фенил, стерически переполненная и электронно -патронная группа: синтез и кислотность пентакиса (трифторметил) бензола, -toluene, -фенол и -анилина». J. Org. Химический 73 (7): 2607–2620. doi : 10.1021/jo702513w . PMID 18324831 .
- ^ Jump up to: а беременный Kütt, A.; Лейто, я.; Калджуранд, я.; Sooväli, L.; Власов, виртуальная машина; Ягупольский, LM; Koppel, IA (2006). «Комплексная самосогласованная спектрофотометрическая шкала кислотности нейтральных кислот Brønsted в ацетонитриле». J. Org. Химический 71 (7): 2829–2838. doi : 10.1021/jo060031y . PMID 16555839 . S2CID 8596886 .
- ^ Калджуранд, я.; Kütt, A.; Sooväli, L.; Родима, Т.; Mäemets, v.; Лейто, я; Koppel, IA (2005). «Расширение самосогласованной шкалы спектрофотометрической базовой основы в ацетонитриле до полного промежутка 28 единиц PKA: объединение различных шкал базовой основы». J. Org. Химический 70 (3): 1019–1028. doi : 10.1021/jo048252w . PMID 15675863 .
- ^ «Бордвелл таблица PKA (кислотность в ДМСО)» . Архивировано из оригинала 9 октября 2008 года . Получено 2008-11-02 .
- ^ Housecroft, CE; Sharpe, AG (2008). Неорганическая химия (3 -е изд.). Прентис Холл. ISBN 978-0-13-175553-6 Полем Глава 8: Не впаживание средств массовой информации
- ^ Rochester, CH (1970). Функции кислотности . Академическая пресса. ISBN 0-12-590850-4 .
- ^ Ола, Джорджия; Prakash, S; Соммер, Дж. (1985). Супероизды . Нью -Йорк: Wiley Interscience. ISBN 0-471-88469-3 .
- ^ Coetzee, JF; Падманабхан, Гр (1965). «Протон-акцепторная власть и гомоконъюгация моно- и диаминов». J. Am. Химический Соц 87 (22): 5005–5010. doi : 10.1021/ja00950a006 .
- ^ Сосновая, Ш; Хендриксон, JB; Крэм, диджей; Hammond, GS (1980). Органическая химия . МакГроу - Хилл. п. 203. ISBN 0-07-050115-7 .
- ^ Box, KJ; Völgyi, G.; Руис, Р.; Comer, Je; Takács-Novák, K.; Bosch, E.; Ràfols, C.; Розес, М. (2007). «Физико -химические свойства новой многокомпонентной системы координата для определения PKA плохо растворимых фармацевтических соединений». Хельв Чим. Акт . 90 (8): 1538–1553. doi : 10.1002/hlca.200790161 .
- ^ Jump up to: а беременный Housecroft, Кэтрин Э.; Шарп, Алан Г. (2005). Неорганическая химия (2 -е изд.). Харлоу, Великобритания: Пирсон Прентис Холл. С. 170–171. ISBN 0-13-039913-2 .
- ^ Jump up to: а беременный Дуглас Б., Макдэниел Д.Х. и Александр Дж.Дж. Концепции и модели неорганической химии (2 -е изд. Wiley 1983) с.526 ISBN 0-471-21984-3
- ^ Полинг Л. (1960). Природа химической связи и структура молекул и кристаллов; Введение в современную структурную химию (3 -е изд.). Итака (Нью -Йорк): издательство Корнелльского университета. п. 277 ISBN 0-8014-0333-2 .
- ^ Сосновая, Ш; Хендриксон, JB; Крэм, диджей; Hammond, GS (1980). Органическая химия . МакГроу - Хилл. ISBN 0-07-050115-7 Полем Раздел 13-3: Количественные корреляции заместительных эффектов (часть B)-уравнение Хамметта
- ^ Hammett, LP (1937). «Влияние структуры на реакции органических соединений. Деривативы бензола». J. Am. Химический Соц 59 (1): 96–103. doi : 10.1021/ja01280a022 .
- ^ Hansch, C.; Лео, А.; Taft, RW (1991). «Обзор констант -заместителей Хамметт и резонанс и параметры поля». Химический Преподобный 91 (2): 165–195. doi : 10.1021/cr00002a004 . S2CID 97583278 .
- ^ Шортер, J (1997). «Компиляция и критическая оценка параметров и уравнений структурной реактивности: часть 2. Расширение шкалы Hammett σ посредством данных для ионизации замещенных бензойных кислот в водных растворителях при 25 ° C (технический отчет)» . Чистая и прикладная химия . 69 (12): 2497–2510. doi : 10.1351/pac199769122497 . S2CID 98814841 .
- ^ Сосновая, Ш; Хендриксон, JB; Крэм, диджей; Hammond, GS (1980). Органическая химия . МакГроу - Хилл. ISBN 0-07-050115-7 Полем Раздел 6-2: Структурное воздействие на кислотность и основность
- ^ Ольха, RW; Боуман, PS; Стил, WRS; Winterman, DR (1968). «Замечательная основность 1,8-бис (диметиламино) нафталина». Химический Общение (13): 723–724. doi : 10.1039/c19680000723 .
- ^ Олдер, RW (1989). «Джайн -эффекты на основные амины». Химический Преподобный 89 (5): 1215–1223. doi : 10.1021/cr00095a015 .
- ^ Аткинс, Питер Уильям; Де Паула, Хулио (2006). Физическая химия Аткинса . Нью -Йорк: WH Freeman. п. 94 ISBN 978-0-7167-7433-4 .
- ^ Мартелл, Ае; Motekaitis, RJ (1992). Определение и использование констант стабильности . Уайли. ISBN 0-471-18817-4 Полем Глава 4: Экспериментальная процедура для потенциометрического измерения рН металлического комплексного равновесия
- ^ Leggett, DJ (1985). Вычислительные методы для определения констант формирования . Пленум. ISBN 0-306-41957-2 .
- ^ Аллен, Ри; Box, KJ; Comer, JEA; Пик, C.; ТАМ, Кен. (1998). «Многоволновое спектрофотометрическое определение константы диссоциации кислоты ионизируемых лекарств». J. Pharm. Биом. Анал 17 (4–5): 699–712. doi : 10.1016/s0731-7085 (98) 00010-7 . PMID 9682153 .
- ^ Box, KJ; Донкор, Re; Юпп, Пенсильвания; Лидер, IP; Trew, df; Тернер, Ч. (2008). «Химия многопрофильных препаратов, часть 1: потенциометрическое, многоволновое титриметрическое исследование PH и титриметрическое исследование PH на лыжных показателях». J. Pharm. Биом. Анал 47 (2): 303–311. doi : 10.1016/j.jpba.2008.01.015 . PMID 18314291 .
- ^ Попов, К.; Ronkkomaki, H.; Lajunen, LHJ (2006). «Руководство по проводникам ЯМР для определения высоких и низких значений PK A » (PDF) . Чистое приложение. Химический 78 (3): 663–675. doi : 10.1351/pac200678030663 . S2CID 4823180 .
- ^ Szakács, Z.; Hägele, G. (2004). "Точное определение низких p k значений 1 ЯМР H титрование (4): 819–825. doi:10.1016/j.talanta.2003.10.007. PMID 18969368.
- ^ Фейг, Эндрю Л., изд. (2016). «Методы в фермере». Калориметрия . 567 . Elsevier: 2–493. ISSN 0076-6879 .
- ^ Jump up to: а беременный в Splittgerber, Ag; Chinander, LL (1 февраля 1988 г.). «Спектр диссоциации промежуточного звена цистеина: эксперимент с биофизической химией». Журнал химического образования . 65 (2): 167. Bibcode : 1988jched..65..167s . doi : 10.1021/ed065p167 .
- ^ Frassineti, C.; Alderighi, L; Ганс, P; Сабатини, а; Вакка, а; Гелли С. (2003). "Определение протониционных констант некоторых фторированных полиаминов с помощью 13 C ЯМР Данные обработаны новой компьютерной программой Hypnmr2000. Последовательность протонирования в полиаминах ». Anal. Bioanal. Chem . 376 (7): 1041–1052. : 10.1007 /S00216-003-2020-0 . PMID 12845401. . S2CID 14533024 DOI
- ^ Onufriev, A.; Дело, да; Ullmann GM (2001). «Новый вид титрования pH в биомолекулах». Биохимия . 40 (12): 3413–3419. doi : 10.1021/bi002740q . PMID 11297406 .
- ^ Хорошо, ne; Вингет, GD; Зима, W.; Коннолли, Теннесси; Izawa, S.; Сингх, RMM (1966). «Ионные буферы водорода для биологических исследований». Биохимия . 5 (2): 467–477. doi : 10.1021/bi00866a011 . PMID 5942950 .
- ^ Данн, MJ (1993). Гелевый электрофорез: белки . Bios Scientific Publishers. ISBN 1-872748-21-x .
- ^ Мартин Р. (1996). Гелевой электрофорез: нуклеиновые кислоты . Bios Scientific Publishers. ISBN 1-872748-28-7 .
- ^ Бреннер, Б.М.; Stein, JH, Eds. (1979). Кислотный базовый и калий гомеостаз . Черчилль Ливингстон. ISBN 0-443-08017-8 .
- ^ Скорпион Р. (2000). Основы кислот, оснований, буферов и их применение в биохимических системах . Kendall/Hunt Pub. Co. ISBN 0-7872-7374-0 .
- ^ Бейнон, RJ; Истерби, JS (1996). Буферные решения: основы . Оксфорд: издательство Оксфордского университета. ISBN 0-19-963442-4 .
- ^ Perrin, DD; Демпси Б. (1974). Буферы для контроля ионов рН и металла . Лондон: Чепмен и Холл. ISBN 0-412-11700-2 .
- ^ Гарфин , Д.; Ahuja, S., eds. (2005). Справочник по изоэлектрической фокусировке и протеомике . Тол. 7. Elsevier. ISBN 0-12-088752-5 .
- ^ Hulanicki, A. (1987). Реакции кислот и оснований в аналитической химии . Массон, MR (редактор перевода). Хорвуд. ISBN 0-85312-330-6 .
- ^ Эяль, А.М. (1997). «Экстракция кислоты с кислотой-базой экстрагтантами». Ионовый обмен и извлечение растворителя: серия достижений . 13 : 31–94.
- ^ Avdeef, A. (2003). Поглощение и разработка лекарств: растворимость, проницаемость и состояние заряда . Нью -Йорк: Уайли. ISBN 0-471-42365-3 .
- ^ Beck, Mt; Нагипал И. (1990). Химия сложных равновесия . Хорвуд. ISBN 0-85312-143-5 .
- ^ Leowen, CJ; Hers, Lem (1995). Оценка риска или химикаты: введение . Спрингер. стр. 254–255. ISBN 0-7923-3740-9 .
- ^ Скуг, да; Запад, DM; Холлер, JF; Crouch, SR (2004). Основы аналитической химии (8 -е изд.). Томсон Брукс/Коул. ISBN 0-03-035523-0 Полем Глава 9-6: кислотный дождь и буферная способность озер
- ^ Stumm, W.; Morgan, JJ (1996). Химия воды . Нью -Йорк: Уайли. ISBN 0-471-05196-9 .
- ^ Snoeyink, VL; Дженкинс, Д. (1980). Водная химия: химические равновесия и скорость в природных водах . Нью -Йорк: Уайли. ISBN 0-471-51185-4 .
- ^ Millero, FJ (2006). Химическая океанография (3 -е изд.). Лондон: Тейлор и Фрэнсис. ISBN 0-8493-2280-4 .
- ^ Милреро, FJ; Лю, X. (2002). «Растворимость железа в морской воде». Морская химия . 77 (1): 43–54. Bibcode : 2002march..77 ... 43L . doi : 10.1016/s0304-4203 (01) 00074-3 .
- ^ Speight, JG (2005). Справочник по химии Ланге (18 -е изд.). МакГроу - Хилл. ISBN 0-07-143220-5 Полем Глава 8
Дальнейшее чтение
[ редактировать ]- Альберт, А.; Serjeant, EP (1971). Определение констант ионизации: лабораторное руководство . Чепмен и Холл. ISBN 0-412-10300-1 Полем (Предыдущее издание опубликовано как Константы ионизации кислот и оснований . Лондон (Великобритания): Метуэн. 1962. )
- Аткинс, PW; Джонс, Л. (2008). Химические принципы: стремление к пониманию (4 -е изд.). WH Freeman. ISBN 978-1-4292-0965-6 .
- Housecroft, CE; Sharpe, AG (2008). Неорганическая химия (3 -е изд.). Прентис Холл. ISBN 978-0-13-175553-6 Полем (Не впальные растворители)
- Hulanicki, A. (1987). Реакции кислот и оснований в аналитической химии . Хорвуд. ISBN 0-85312-330-6 Полем (Редактор перевода: Мэри Р. Массон)
- Perrin, DD; Демпси, Б.; Serjeant, EP (1981). Прогнозирование PKA для органических кислот и оснований . Чепмен и Холл. ISBN 0-412-22190-х .
- Reichardt, C. (2003). Растворители и эффекты растворителя в органической химии (3 -е изд.). Wiley-Vch. ISBN 3-527-30618-8 Полем Глава 4: Воздействие растворителя на положение гомогенных химических равновесия.
- Скуг, да; Запад, DM; Холлер, JF; Crouch, SR (2004). Основы аналитической химии (8 -е изд.). Томсон Брукс/Коул. ISBN 0-03-035523-0 .
Внешние ссылки
[ редактировать ]- Данные о кислотности и базистости в неакезных растворителях обширная библиография значений P a A в DMSO , ацетонитриле , THF , гептане , 1,2-дихлорэтане и в газовой фазе
- Кертипот все в одном. Основное программное обеспечение для расчетов равновесия с рН и кислотой базы, а также для моделирования и анализа потенциометрических кривых титрования с электронными таблицами
- химического свойства SPARC водной, невой и газообразной фазой P k a Калькулятор физического / включает в себя базу данных с
- Константы водного равновесия p a Значения для различных кислот и оснований. Включает таблицу некоторых продуктов растворимости
- руководство по a и log Интерпретация и p измерение Бесплатное P k
- Бесплатный онлайн -инструмент прогнозирования (Marvin) P K A , Log P , Log D и т. Д. Из Chemaxon
- Chemicalize.org : Список прогнозируемых структурных свойств
- P K диаграмма А. [1] Дэвида Эванса