Константа диссоциации кислоты

В химии — константа диссоциации кислоты (также известная как константа кислотности или константа ионизации кислоты ; обозначается ) . ) количественная мера силы кислоты — в растворе . Это константа равновесия химической реакции.


известный как диссоциация в контексте кислотно-основных реакций . Химическая форма ГК представляет собой кислоту , которая диссоциирует на А − , называемое сопряженным основанием кислоты, и ионом водорода , ЧАС + . [ а ] Говорят, что система находится в равновесии, если концентрации ее компонентов не меняются с течением времени, поскольку как прямая, так и обратная реакции протекают с одинаковой скоростью. [ 1 ]
Константа диссоциации определяется выражением [ б ]
- или по его логарифмической форме
![{\displaystyle K_{\text{a}}=\mathrm {\frac {[A^{-}][H^{+}]}{[HA]}},}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7cdd9efda0e3a32060020b5c9e5b2c78981b2a93)
![{\displaystyle \mathrm {p} K_{{\ce {a}}}=-\log _{10}K_{\text{a}}=\log _{10}{\frac {{\ce {[ HA]}}}{[{\what {A^-}}][{\what {H+}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d7af05bf129db2f9bc618fe809660b6e4ff8dce9)
где количества в квадратных скобках представляют собой молярные концентрации веществ в равновесии. [ с ] [ 2 ] Например, гипотетическая слабая кислота, имеющая K a = 10 −5 значение log K a представляет собой показатель степени (−5), что дает p K a = 5. Для уксусной кислоты K a = 1,8 x 10 −5 , поэтому p K a составляет около 5. Более высокий K a соответствует более сильной кислоте (кислоте, которая более диссоциирует в равновесии). Форма pKa часто используется , поскольку она обеспечивает удобную логарифмическую шкалу , где более низкое значение соответствует pKa более сильной кислоте.
Теоретическая основа
[ редактировать ]Константа диссоциации кислоты является прямым следствием основной термодинамики реакции диссоциации; значение p K a прямо пропорционально стандартному изменению свободной энергии Гиббса для реакции. Значение p K a меняется с температурой и качественно может быть понято на основе принципа Ле Шателье : когда реакция эндотермическая , K a увеличивается, а p K a уменьшается с увеличением температуры; обратное верно для экзотермических реакций.
Величина p K a также во многом зависит от молекулярной структуры кислоты. Например, Полинг предложил два правила: одно для последовательных p K a полипротонных кислот (см. Полипротонные кислоты ниже), а другое для оценки p K a оксикислот на основе количества групп = O и -OH (см. Факторы, влияющие на p K a значения ниже). Другие структурные факторы, влияющие на величину константы диссоциации кислоты, включают индуктивные эффекты , мезомерные эффекты и водородную связь . Уравнения типа Хэммета часто применялись для оценки p K a . [ 3 ] [ 4 ]
Количественное поведение кислот и оснований в растворах можно понять, только если рКа их известны значения . В частности, pH раствора можно предсказать, если p K a известны аналитическая концентрация и значения всех кислот и оснований; и наоборот, если известен pH, можно рассчитать равновесную концентрацию кислот и оснований в растворе. Эти расчеты находят применение во многих различных областях химии, биологии, медицины и геологии. Например, многие соединения, используемые в лекарствах, представляют собой слабые кислоты или основания, и знание значений p K a вместе с коэффициентом распределения октанол-вода может быть использовано для оценки степени, в которой соединение попадает в кровоток. Константы диссоциации кислоты также важны в водной химии и химической океанографии , где кислотность воды играет фундаментальную роль. В живых организмах кислотно-основной гомеостаз и кинетика ферментов зависят от значений p K a многих кислот и оснований, присутствующих в клетке и организме. По химии знание п. Значения K a необходимы для приготовления буферных растворов , а также являются предпосылкой для количественного понимания взаимодействия кислот или оснований с ионами металлов с образованием комплексов . Экспериментально значения p K a могут быть определены потенциометрическим (pH) титрованием , но для значений p K a менее примерно 2 или более примерно 11 спектрофотометрические измерения или измерения ЯМР могут потребоваться из-за практических трудностей с измерениями pH.
Определения
[ редактировать ]Согласно Аррениуса первоначальному молекулярному определению , кислота — это вещество, которое диссоциирует в водном растворе, выделяя ион водорода. ЧАС + (протон): [ 5 ]

Константа равновесия этой реакции диссоциации известна как константа диссоциации . Освободившийся протон соединяется с молекулой воды, образуя ион гидроксония (или оксония). Н 3 О + (голые протоны в растворе не существуют), поэтому позже Аррениус предложил записать диссоциацию как кислотно-основную реакцию :

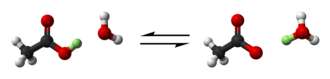
Брёнстед и Лоури обобщили это на реакцию протонного обмена: [ 6 ] [ 7 ] [ 8 ]

Кислота теряет протон, оставляя сопряженное основание; протон переносится на основание, образуя сопряженную кислоту. Для водных растворов кислоты ГК основанием является вода; сопряженное основание А − и сопряженная кислота представляет собой ион гидроксония. Определение Бренстеда-Лоури применимо к другим растворителям, таким как диметилсульфоксид : растворитель S действует как основание, принимая протон и образуя сопряженную кислоту. Ш + .

В химии растворов обычно используют ЧАС + как сокращение для сольватированного иона водорода, независимо от растворителя. В водном растворе ЧАС + обозначает сольватированный ион гидроксония, а не протон. [ 9 ] [ 10 ]
Обозначение кислоты или основания как «конъюгата» зависит от контекста. Сопряженная кислота ЧД + основания B диссоциирует по

что является противоположностью равновесия

Гидроксид -ион ОЙ − , хорошо известное основание, действует здесь как сопряженное основание кислой воды. Таким образом, кислоты и основания рассматриваются просто как доноры и акцепторы протонов соответственно.
Более широкое определение диссоциации кислоты включает гидролиз , при котором протоны образуются в результате расщепления молекул воды. Например, борная кислота ( B(OH) 3 ) производит Н 3 О + как если бы это был донор протонов, [ 11 ] было подтверждено но с помощью рамановской спектроскопии , что это связано с равновесием гидролиза: [ 12 ]

Аналогично, гидролиз ионов металлов приводит к образованию таких ионов, как [Ал(Н 2 О) 6 ] 3+ вести себя как слабые кислоты: [ 13 ]
![{\displaystyle {\ce {[Al(H2O)6]^3+ + H2O <=> [Al(H2O)5(OH)]^2+ + H3O+}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b1c60923d504a87f8bbd22293ac8eaad8341ea41)
Согласно первоначальному определению Льюиса , кислота — это вещество, которое принимает электронную пару с образованием координатной ковалентной связи . [ 14 ]
Константа равновесия
[ редактировать ]Константа диссоциации кислоты является частным примером константы равновесия . Диссоциацию монопротонной кислоты ГК в разбавленном растворе можно записать как
Константа термодинамического равновесия может быть определен как [ 15 ]


где представляет активность в равновесии химического соединения X. безразмерен , поскольку деятельность безразмерна. В числителе стоят активности продуктов диссоциации, в знаменателе – активности реагирующих веществ. См. коэффициент активности для получения этого выражения.


Поскольку активность является продуктом концентрации и коэффициента активности ( γ ), определение также можно записать как
![{\displaystyle K^{\ominus }={{\frac {[{\ce {A^-}}][{\ce {H+}}]}{{\ce {[HA]}}}}\Gamma },\quad \Gamma ={\frac {\gamma _{{\ce {A^-}}}\ \gamma _{{\ce {H+}}}}}{\gamma _{{\ce {HA } }}\ }}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6e9373db7091aeb4f51a26757a677b420f0a8418)
где представляет собой концентрацию HA и — частное коэффициентов активности.
![{\displaystyle [{\text{HA}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3cfe8305c0735d25de8cef20edf09ef5144d700a)

Во избежание осложнений, связанных с использованием активностей, константы диссоциации определяют , по возможности, в среде высокой ионной силы , то есть в условиях, в которых можно считать всегда постоянным. [ 15 ] Например, среда может представлять собой раствор 0,1 молярного (М) нитрата натрия или 3 М перхлората калия . При этом предположении,
![{\displaystyle K_{\text{a}}={\frac {K^{\ominus }}{\Gamma }}=\mathrm {\frac {[A^{-}][H^{+}]} {[ХА]}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/2a5a59c740de89347ec4c96d982292fc05c64b2f)
![{\displaystyle \mathrm {p} K_{{\ce {a}}}=-\log _{10}{\frac {[{\ce {A^-}}][{\ce {H^+} }]}{[{\ce {HA}}]}}=\log _{10}{\frac {{\ce {[HA]}}}{[{\ce {A^-}}][{ \что {H+}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/bed5fbab82167a42994a6d735931d08b06f1e7a5)
получается. Однако обратите внимание, что все опубликованные значения констант диссоциации относятся к конкретной ионной среде, используемой при их определении, и что разные значения получаются в разных условиях, как показано для уксусной кислоты на иллюстрации выше. Когда опубликованные константы относятся к ионной силе, отличной от той, которая требуется для конкретного применения, они могут быть скорректированы с помощью специфической теории ионов (SIT) и других теорий. [ 16 ]
Кумулятивные и ступенчатые константы
[ редактировать ]Кумулятивная константа равновесия, обозначаемая связан с произведением ступенчатых констант, обозначаемым Для двухосновной кислоты соотношение между ступенчатыми и общими константами следующее:



![{\displaystyle \beta _{2}={\frac {{\ce {[H2A]}}}{[{\ce {A^2-}}][{\ce {H+}}]^{2} }}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/08598ffd39aa7af9e4d7ca73764ada00fdc0882f)

Обратите внимание, что в контексте образования комплексов металл-лиганд константы равновесия для образования металлокомплексов обычно определяются как константы ассоциации . В этом случае константы равновесия протонирования лиганда также определяются как константы ассоциации. Нумерация констант ассоциации обратна нумерации констант диссоциации; в этом примере

Константы ассоциации и диссоциации
[ редактировать ]При обсуждении свойств кислот константы равновесия обычно определяют как константы диссоциации кислот, обозначаемые K a , с числовыми значениями, обозначаемыми символом p K a .
![{\displaystyle K_{\text{dissoc}}={\frac {{\ce {[A- ][H+]}}}{{\ce {[HA]}}}}:\mathrm {p} K_{ \text{a}}=-\log K_{\text{dissoc}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c115ab88c5f847b2fe5c3250d9e5c9134d125080)
С другой стороны, для оснований используются константы ассоциации.
![{\displaystyle K_{\text{assoc}}={\frac {{\ce {[HA]}}}{{\ce {[A- ][H+]}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e353beeaa76919ab646b6969e6d30f7e01fe7afa)
Однако компьютерные программы общего назначения , которые используются для получения значений констант равновесия на основе экспериментальных данных, используют константы ассоциации как для кислот, так и для оснований. Поскольку константы стабильности комплекса металл-лиганд всегда указываются как константы ассоциации, протонирование лиганда также необходимо указывать как реакцию ассоциации. [ 17 ] Определения показывают, что значение константы диссоциации кислоты обратно пропорционально значению соответствующей константы ассоциации:



Примечания
- Для данной кислоты или основания в воде p K a + p K b = p K w , константа самоионизации воды .
- Константу ассоциации образования супрамолекулярного комплекса можно обозначить как K a ; в таких случаях «а» означает «ассоциацию», а не «кислоту».
- Для полипротонных кислот нумерация ступенчатых констант ассоциации обратна нумерации констант диссоциации. Например, для фосфорной кислоты (подробности см. в разделе « Полипротонные кислоты» ниже):

Температурная зависимость
[ редактировать ]Все константы равновесия изменяются с температурой в соответствии с уравнением Ван 'т-Гоффа. [ 18 ]

— газовая постоянная , — абсолютная температура . Таким образом, для экзотермических реакций стандартное изменение энтальпии , , является отрицательным, а K уменьшается с температурой. Для эндотермических реакций положителен, и K увеличивается с температурой.



Стандартное изменение энтальпии реакции само по себе является функцией температуры в соответствии с законом термохимии Кирхгофа :

где — изменение теплоемкости при постоянном давлении. На практике можно считать постоянным в небольшом диапазоне температур.

Размерность
[ редактировать ]В уравнении
![{\displaystyle K_{\mathrm {a} }=\mathrm {\frac {[A^{-}][H^{+}]}{[HA]}} ,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/441ece0dee32e0a14fe14d4b1678785804486a92)
K a, по-видимому, имеет размерность концентрации. Однако, поскольку , константа равновесия, , не может иметь физического измерения. Этот кажущийся парадокс можно разрешить разными способами.


- Предположим, что частное коэффициентов активности имеет числовое значение 1, так что имеет то же численное значение, что и константа термодинамического равновесия. .
- Выразите каждое значение концентрации как соотношение c/c 0 , где с 0 — это концентрация в [гипотетическом] стандартном состоянии с числовым значением 1 по определению. [ 19 ]
- Выразите концентрации в шкале мольных фракций . Поскольку мольная доля не имеет размерности, частное концентраций по определению будет чистым числом.
Процедуры (1) и (2) дают одинаковые численные значения константы равновесия. Более того, поскольку концентрация просто пропорционален мольной доле и плотность :




и поскольку молярная масса — константа в разбавленных растворах, значение константы равновесия, определенное с помощью (3), будет просто пропорционально значениям, полученным с помощью (1) и (2).

принято В биохимии указывать значение с размером, например, « K a = 30 мМ», чтобы указать масштаб, миллимолярный (мМ) или микромолярный (мкМ) значений концентрации , используемых для его расчета.
Сильные кислоты и основания
[ редактировать ]Кислота классифицируется как «сильная», если концентрация ее недиссоциированных частиц слишком мала для измерения. [ 6 ] Любая водная кислота со значением apK 0 меньше практически полностью депротонирована и считается сильной кислотой . [ 20 ] Все такие кислоты передают свои протоны воде и образуют катионы растворителя (H 3 O + в водном растворе), так что все они имеют по существу одинаковую кислотность — явление, известное как выравнивание растворителя . [ 21 ] [ 22 ] Говорят, что они полностью диссоциированы в водном растворе, поскольку количество недиссоциировавшей кислоты, находящейся в равновесии с продуктами диссоциации, ниже предела обнаружения . Аналогично, любое водное основание с константой ассоциации p K b менее примерно 0, что соответствует p K a более примерно 14, выравнивается до OH. − и считается сильной базой . [ 22 ]
Азотная кислота со значением ap K около -1,7 ведет себя как сильная кислота в водных растворах с pH более 1. [ 23 ] При более низких значениях pH он ведет себя как слабая кислота.
Значения p K a для сильных кислот были оценены теоретическим путем. [ 24 ] Например, значение p K a водного раствора HCl оценивается как -9,3.
Монопротонные кислоты
[ редактировать ]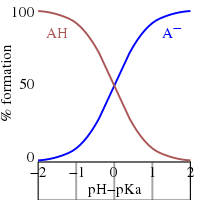
После перестановки выражения, определяющего K a , и помещения pH = -log 10 [H + ] , получаем [ 25 ]
![{\displaystyle \mathrm {pH} =\mathrm {p} K_ {\text{a}}+\log \mathrm {\frac {[A^{-}]}{[HA]}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/25e874f2b8ea8e4127605788c356393cfd7fff37)
Это уравнение Хендерсона–Хассельбаха , из которого можно сделать следующие выводы.
- При полунейтрализации соотношение [А − ] / [ГА] знак равно 1 ; поскольку log(1) = 0 , pH при полунейтрализации численно равен p K a . И наоборот, при pH = p K a концентрация ГК равна концентрации А. − .
- Буферная область простирается в приблизительном диапазоне p K a ± 2. Буферизация слаба за пределами диапазона p K a ± 1. При pH ≤ p K a − 2 вещество считается полностью протонированным, а при pH ≥ p K a + 2 полностью диссоциирован (депротонирован).
- Если известен pH, можно рассчитать соотношение. Это соотношение не зависит от аналитической концентрации кислоты.
В воде измеримые значения p K a варьируются от примерно -2 для сильной кислоты до примерно 12 для очень слабой кислоты (или сильного основания).
Буферный раствор с желаемым pH можно приготовить как смесь слабой кислоты и сопряженного с ней основания. На практике смесь можно создать, растворив кислоту в воде и добавив необходимое количество сильной кислоты или основания. Когда известны p K a и аналитическая концентрация кислоты, степень диссоциации и pH раствора монопротонной кислоты можно легко рассчитать с помощью таблицы ICE .
Полипротонные кислоты
[ редактировать ]
Полипротонная кислота — это соединение, которое может потерять более 1 протона. Каждая константа ступенчатой диссоциации определяется для потери одного протона. Константу диссоциации первого протона можно обозначить как К а1 , а константы диссоциации последующих протонов — как К а2 и т. д. Фосфорная кислота H 3 PO 4 является примером полипротонной кислоты, поскольку она может терять три протона.
Равновесие p K определение и значение [ 26 ]

![{\displaystyle \mathrm {p} K_{{\ce {a1}}}=\log _{10}{\frac {[{\ce {H_3PO_4}}]}{[{\ce {H_2PO_4^{-} }}][{\ce {H^+}}]}}=2,14}](https://wikimedia.org/api/rest_v1/media/math/render/svg/dff6e67381cb8a691b8873fbf884dad30b001352)

![{\displaystyle \mathrm {p} K_{{\ce {a2}}}=\log _{10}{\frac {[{\ce {H_2PO_4^{-}}}]}{[{\ce {HPO_4 ^{2-}}}][{\what {H^+}}]}}=7.2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/efe9f5a620a62c8de4a6567f58faf01e66829903)

![{\displaystyle \mathrm {p} K_{{\ce {a3}}}=\log _{10}{\frac {[{\ce {HPO4^2-}}]}{[{\ce {PO4^ 3-}}][{\ce {H+}}]}}=12,37}](https://wikimedia.org/api/rest_v1/media/math/render/svg/dfac57be595c190a1dc60479c2538e575488de02)
Когда разница между последовательными значениями p K составляет около четырех или более, как в этом примере, каждый вид можно рассматривать как кислоту сам по себе; [ 27 ] Фактически соли H
2 ПО −
4 может быть кристаллизован из раствора путем доведения pH примерно до 5,5 и солей HPO 2- 4 можно кристаллизовать из раствора, доведя pH примерно до 10. Диаграмма распределения видов показывает, что концентрации двух ионов максимальны при pH 5,5 и 10.
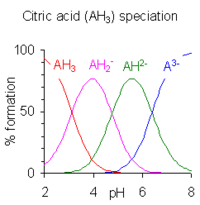
Когда разница между последовательными значениями p K меньше примерно четырех, происходит перекрытие диапазонов pH существования видов в равновесии. Чем меньше разница, тем больше совпадение. Случай лимонной кислоты показан справа; растворы лимонной кислоты забуферены во всем диапазоне pH от 2,5 до 7,5.
Согласно первому правилу Полинга, последовательные значения p K данной кислоты увеличиваются (p K a2 > p K a1 ) . [ 28 ] Для оксикислот с более чем одним ионизируемым водородом на одном и том же атоме значения p K a часто увеличиваются примерно на 5 единиц на каждый удаленный протон, [ 29 ] [ 30 ] как в примере с фосфорной кислотой выше.
Из таблицы выше видно, что второй протон удаляется из отрицательно заряженной частицы. Поскольку протон несет положительный заряд, для его удаления необходима дополнительная работа, поэтому p K a2 больше, чем p K a1 . p K a3 больше, чем p K a2 , поскольку происходит дальнейшее разделение зарядов. Когда обнаруживается исключение из правила Полинга, это указывает на то, что также происходят серьезные изменения в структуре. В случае VO + 2 (водн.), ванадий октаэдрический , 6-координатный, тогда как ванадиевая кислота тетраэдрический , 4-координатный. Это означает, что при первой диссоциации высвобождаются четыре «частицы», но при других диссоциациях высвобождаются только две «частицы», что приводит к гораздо большему вкладу энтропии в стандартное изменение свободной энергии Гиббса для первой реакции, чем для остальных.
Равновесие п К а
![{\displaystyle {\ce {[VO2(H2O)4]+ <=> H3VO4 + H+ + 2H2O}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0b5fcdc28e4fbd98292ebd608643c231844a334f)







Изоэлектрическая точка
[ редактировать ]Для веществ в растворе изоэлектрическая точка (p I ) определяется как pH, при котором взвешенная по величине заряда сумма концентраций положительно заряженных частиц равна взвешенной сумме концентраций отрицательно заряженных частиц. В случае, если имеется по одному виду каждого типа, изоэлектрическую точку можно получить непосредственно из p K. значений Возьмем, к примеру, глицин , определяемый как AH. Следует рассмотреть два равновесия диссоциации.
![{\displaystyle {\ce {AH2+<=>AH~+H+\qquad [AH][H+]={\mathit {K}}_{1}[AH2+]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/359f1d34ddc5ac4b4cecf45e11539bc14462e98f)
![{\displaystyle {\ce {AH<=>A^{-}~+H+\qquad [A^{-}][H+]={\mathit {K}}_{2}[AH]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/95c1af1bef047675675659473e3886aefff9fa79)
Подставим выражение для [AH] из второго уравнения в первое уравнение.
![{\displaystyle {\ce {[A^{-}][H+]^{2}={\mathit {K}}_{1}{\mathit {K}}_{2}[AH2+]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/473772e02e925c83975026ce31b43a0a2dc4b1cc)
В изоэлектрической точке концентрация положительно заряженных частиц AH + 2 равен концентрации отрицательно заряженных частиц, А − , так
![{\displaystyle [{\ce {H+}}]^{2}=K_{1}K_{2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3dfa9985f2d6769f35f98f98153ac6cabd9c011b)
Следовательно, если принять кологарифмы , pH определяется выражением

Значения p I для аминокислот указаны для протеиногенных аминокислот . Когда более двух заряженных видов находятся в равновесии друг с другом, может потребоваться полный расчет видообразования.
Основы и основность
[ редактировать ]Константа равновесия K b для основания обычно определяется как константа ассоциации протонирования основания B с образованием сопряженной кислоты: полупансион + .

Используя рассуждения, аналогичные использованным ранее
![{\displaystyle {\begin{aligned}K_{\text{b}}&=\mathrm {\frac {[HB^{+}][OH^{-}]}{[B]}} \\\mathrm {p} K_{\text{b}}&=-\log _{10}\left(K_{\text{b}}\right)\end{aligned}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5dea1aac629a595476e18c042a8f4365a50f0efc)
K b связан с K a для сопряженной кислоты. В воде концентрация гидроксид- иона, [ОЙ − ] , связано с концентрацией иона водорода соотношением К ш = [Н + ][ОЙ − ] , поэтому
![{\displaystyle \mathrm {[OH^{-}]} = {\frac {K_ {\mathrm {w} }}{\ mathrm {[H^{+}]} }}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ab7f583da9f8b50145990ffa4342919930edfa16)
Замена выражения на [ОЙ − ] в выражение для K b дает
![{\displaystyle K_{\text{b}}={\frac {[\mathrm {HB^{+}} ]K_{\text{w}}}{\mathrm {[B][H^{+}] } }}={\frac {K_{\text{w}}}{K_{\text{a}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/921c3abd37a1c5c00c31831509d3b090394c0d47)
Когда K a , K b и K w определяются при одинаковых условиях температуры и ионной силы, то, принимая кологарифмы , следует, что p K b = p K w − p K a . В водных растворах при 25 °C p K w составляет 13,9965, [ 31 ] так

с достаточной точностью для большинства практических целей. Фактически нет необходимости определять p K b отдельно от p K a , [ 32 ] но здесь это сделано, так как p K b в старой литературе часто можно найти только значения .
Для гидролизованного иона металла K b также можно определить как ступенчатой диссоциации. константу

![{\displaystyle K_{\mathrm {b} }={\frac {[\mathrm {M} _{p}({\ce {OH}})_{q-1}^{+}][{\ce {OH-}}]}{[\mathrm {M} _{p}({\ce {OH}})_{q}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8391968e700e57f56f6041dd839f0fdb5e93780e)
Это обратная константа ассоциации для образования комплекса.
Основность выражается как константа диссоциации сопряженной кислоты.
[ редактировать ]Поскольку соотношение p K b = p K w - p K a справедливо только в водных растворах (хотя аналогичные соотношения применимы и для других амфотерных растворителей), в таких разделах химии, как органическая химия , которые обычно имеют дело с неводными растворами, обычно не используется p K b в качестве мера основности. Вместо этого, p K a сопряженной кислоты, обозначаемой p K aH , указывается, когда необходимо количественно определить основность. Для основания B и сопряженной с ним кислоты BH + в равновесии это определяется как
![{\ displaystyle \ mathrm {p} K_ {\ mathrm {aH} } (\ mathrm {B}) = \ mathrm {p} K_ {\ mathrm {a} } ({\ ce {BH+}}) = - \ log _{10}{\Big (}{\frac {[{\ce {B}}][{\ce {H+}}]}{[{\ce {BH+}}]}}{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b76149bc3dbc3d0375d6355bdf2342394a568776)
Более высокое значение p K aH соответствует более сильному основанию. Например, значения p K aH (C 5 H 5 N) = 5,25 и p K aH ((CH 3 CH 2 ) 3 N) = 10,75 указывают на то, что (CH 3 CH 2 ) 3 N (триэтиламин) является более сильным основанием, чем C 5 H 5 N (пиридин).
Амфотерные вещества
[ редактировать ]Амфотерное вещество – это вещество, которое в зависимости от pH может действовать как кислота или как основание. Вода (внизу) амфотерна. Другим примером амфотерной молекулы является бикарбоната . ион HCO − 3 , которое является сопряженным основанием молекулы угольной кислоты H2CO3 в равновесии

но также и кислота, сопряженная с карбонат- ионом CO 2− 3 в (обратном) равновесии

Равновесие углекислоты важно для кислотно-основного гомеостаза в организме человека.
Аминокислота также является амфотерной , однако ее нейтральная молекула находится в состоянии внутреннего кислотно-щелочного равновесия, при котором основная аминогруппа притягивает и связывает протон кислой карбоксильной группы, образуя цвиттер-ион .

При pH менее примерно 5 протонируются как карбоксилатная группа, так и аминогруппа. По мере увеличения pH кислота диссоциирует по

При высоком pH может иметь место вторая диссоциация.

Таким образом, молекула аминокислоты является амфотерной, поскольку она может быть как протонирована, так и депротонирована.
Самоионизация воды
[ редактировать ]Молекула воды может как приобрести, так и потерять протон. Говорят, что он амфипротный . Ионизационное равновесие можно записать

где в водном растворе ЧАС + обозначает сольватированный протон. Часто это записывают как гидроксония. ион Н 3 О + , но эта формула не точна, поскольку на самом деле происходит сольватация более чем одной молекулой воды и такими видами, как Н 5 О + 2 , H 7 O + 3 и H 9 O + 4 также присутствуют. [ 33 ]
Константа равновесия определяется выражением
![{\displaystyle K_{\text{a}}=\mathrm {\frac {[H^{+}][OH^{-}]}{[H_{2}O]}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/cdc540ad193c8f1661c1897698be93153fc5fb84)
В растворах, в которых концентрации растворенных веществ не очень высоки, концентрация [H 2 O] можно считать постоянным, независимо от растворенного(ых) вещества(й); это выражение затем можно заменить на
![{\displaystyle K_{\text{w}}=[\mathrm {H} ^{+}][\mathrm {OH} ^{-}]\,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c0039f77db244ea2f6d03d3475dc7a232a8ccb16)
константа самоионизации воды Kw Таким образом , является частным случаем константы диссоциации кислоты. Логарифмическая форма, аналогичная p K a, также может быть определена

| Т (°С) | 0 | 5 | 10 | 15 | 20 | 25 | 30 | 35 | 40 | 45 | 50 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| п К ш | 14.943 | 14.734 | 14.535 | 14.346 | 14.167 | 13.997 | 13.830 | 13.680 | 13.535 | 13.396 | 13.262 |
Эти данные можно смоделировать параболой с

Из этого уравнения p K w = 14 при 24,87 °C. При этой температуре ионы водорода и гидроксида имеют концентрацию 10 −7 М.
Кислотность в неводных растворах
[ редактировать ]Растворитель с большей вероятностью будет способствовать ионизации растворенной молекулы кислоты в следующих случаях: [ 35 ]
- Это протонный растворитель , способный образовывать водородные связи.
- У него большое число доноров , что делает его сильной базой Льюиса .
- Он имеет высокую диэлектрическую проницаемость (относительную диэлектрическую проницаемость), что делает его хорошим растворителем ионных частиц.
Значения p K a органических соединений часто получают с использованием апротонных растворителей диметилсульфоксида (ДМСО). [ 35 ] и ацетонитрил (ACN). [ 36 ]
| Растворитель | Номер донора [ 35 ] | Диэлектрическая проницаемость [ 35 ] |
|---|---|---|
| Ацетонитрил | 14 | 37 |
| Диметилсульфоксид | 30 | 47 |
| Вода | 18 | 78 |
растворяет неполярные гидрофобные ДМСО широко используется в качестве альтернативы воде, поскольку он имеет более низкую диэлектрическую проницаемость, чем вода, менее полярен и поэтому легче вещества. Он имеет измеримый pK диапазон от 1 до 30. Ацетонитрил менее щелочной, чем ДМСО, поэтому в этом растворителе обычно кислоты слабее, а основания сильнее. Некоторые pKa значения при 25 °C для ацетонитрила (ACN) [ 37 ] [ 38 ] [ 39 ] и диметилсульфоксид (ДМСО). [ 40 ] показаны в следующих таблицах. Значения для воды включены для сравнения.
| ЕСЛИ ⇌ А − + Ч + | АКС | ДМСО | Вода |
|---|---|---|---|
| п -толуолсульфоновая кислота | 8.5 | 0.9 | Сильный |
| 2,4-динитрофенол | 16.66 | 5.1 | 3.9 |
| Бензойная кислота | 21.51 | 11.1 | 4.2 |
| Уксусная кислота | 23.51 | 12.6 | 4.756 |
| Фенол | 29.14 | 18.0 | 9.99 |
| ЧД + ⇌ Б + Ч + | АКС | ДМСО | Вода |
| Пирролидин | 19.56 | 10.8 | 11.4 |
| Триэтиламин | 18.82 | 9.0 | 10.72 |
| Протонная губка | 18.62 | 7.5 | 12.1 |
| Пиридин | 12.53 | 3.4 | 5.2 |
| Анилин | 10.62 | 3.6 | 4.6 |
Ионизация кислот в кислом растворителе меньше, чем в воде. Например, хлористый водород является слабой кислотой при растворении в уксусной кислоте . Это связано с тем, что уксусная кислота является гораздо более слабым основанием, чем вода.

Сравните эту реакцию с тем, что происходит при растворении уксусной кислоты в более кислом растворителе, чистой серной кислоте: [ 41 ]


Маловероятный геминального диола вид CH 3 C(OH) +2 стабилен в этих средах. Для водных растворов является шкала pH наиболее удобной функцией кислотности . [ 42 ] Другие функции кислотности были предложены для неводных сред, наиболее примечательной из которых является кислотности Гаммета функция H 0 для сверхкислотных сред и ее модифицированная версия H - для сверхосновных сред. [ 43 ]
В апротонных растворителях олигомеры , такие как хорошо известный димер уксусной кислоты , могут образовываться за счет водородных связей. Кислота также может образовывать водородные связи со своим сопряженным основанием. Этот процесс, известный как гомоконъюгация , приводит к повышению кислотности кислот, снижению их эффективных значений p K a за счет стабилизации основания конъюгата. Гомоконъюгация увеличивает протондонорную способность толуолсульфоновой кислоты в растворе ацетонитрила почти в 800 раз. [ 44 ]
В водных растворах гомоконъюгация не происходит, поскольку вода образует более прочные водородные связи с сопряженным основанием, чем кислота.
Смешанные растворители
[ редактировать ]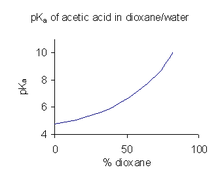
Когда соединение имеет ограниченную растворимость в воде, обычной практикой (например, в фармацевтической промышленности) является определение значений p K a в смеси растворителей, такой как вода/ диоксан или вода/ метанол , в которой соединение более растворимо. [ 46 ] В примере, показанном справа, значение p K a резко возрастает с увеличением процентного содержания диоксана, поскольку диэлектрическая проницаемость смеси уменьшается.
A p K a, Значение полученное в смешанном растворителе, нельзя использовать непосредственно для водных растворов. Причина этого в том, что когда растворитель находится в стандартном состоянии, его активность определяется как единица. Например, стандартное состояние смеси воды и диоксана с соотношением смешивания 9:1 представляет собой именно смесь растворителей без добавления растворенных веществ. Чтобы получить значение pKa использования для с водными растворами, его необходимо экстраполировать до нулевой концентрации сорастворителя из значений, полученных для различных смесей сорастворителей.
Эти факты затенены отсутствием растворителя в выражении, которое обычно используется для определения pKa но , значения , pKa полученные в данном смешанном растворителе, можно сравнивать друг с другом, получая относительную силу кислоты. То же самое справедливо и для значений pKa полученных , в конкретном неводном растворителе, таком как ДМСО.
Универсальная, независимая от растворителя шкала констант диссоциации кислот не разработана, поскольку не известно способа сравнения стандартных состояний двух разных растворителей.
Факторы, влияющие на p K a значения
[ редактировать ]Второе правило Полинга состоит в том, что значение первого p K a для кислот формулы XO m (OH) n зависит прежде всего от числа оксогрупп m и примерно не зависит от количества гидроксильных групп n , а также от количества центральный атом X. Приблизительные значения p K a равны 8 для m = 0, 2 для m = 1, −3 для m = 2 и < −10 для m = 3. [ 28 ] В качестве альтернативы были предложены различные числовые формулы, включая p K a = 8–5 м (известные как правило Белла ), [ 29 ] [ 47 ] п K а знак равно 7 - 5 м , [ 30 ] [ 48 ] или п K а знак равно 9 - 7 м . [ 29 ] Зависимость от m коррелирует со степенью окисления центрального атома X: чем выше степень окисления, тем сильнее оксикислота.
Например, p K a для HClO равно 7,2, для HClO 2 равно 2,0, для HClO 3 равно -1 и HClO 4 является сильной кислотой ( p K a ≪ 0 ). [ 7 ] Повышенная кислотность при добавлении оксогруппы обусловлена стабилизацией сопряженного основания за счет делокализации его отрицательного заряда по дополнительному атому кислорода. [ 47 ] Это правило может помочь определить молекулярную структуру: например, фосфористая кислота , имеющая молекулярную формулу H 3 PO 3 , имеет ap K a около 2, что позволило предположить, что структура представляет собой HPO(OH) 2 , что позднее было подтверждено методом ЯМР-спектроскопии , а не P(OH) 3 , который, как ожидается, будет иметь ap K a около 8. [ 48 ]

Индуктивные эффекты и мезомерные эффекты влияют на значения p K a . Простой пример — эффект замены атомов водорода в уксусной кислоте более электроотрицательным атомом хлора. Электроноакцепторный эффект заместителя облегчает ионизацию, поэтому последовательные значения p K a уменьшаются в ряду 4,7, 2,8, 1,4 и 0,7 при наличии 0, 1, 2 или 3 атомов хлора. [ 49 ] Уравнение Гаммета дает общее выражение влияния заместителей. [ 50 ]
- журнал( К а ) = журнал( К 0
а ) + пс.
K a – константа диссоциации замещенного соединения, K 0
a — константа диссоциации, когда заместителем является водород, ρ — свойство незамещенного соединения, а σ имеет особое значение для каждого заместителя. График зависимости log( K a ) от σ представляет собой прямую линию с пересечением log( K 0
а ) и наклон ρ. Это пример линейной зависимости свободной энергии поскольку log( Ka , ) пропорционален стандартному изменению свободной энергии. Хэммет изначально [ 51 ] связь с данными по бензойной кислоте с различными заместителями в орто- и сформулировал пара - положениях: некоторые числовые значения находятся в уравнении Гаммета . Это и другие исследования позволили упорядочить заместители в зависимости от их электроноакцепторной или электроноотдающей способности, а также различить индуктивные и мезомерные эффекты. [ 52 ] [ 53 ]
Спирты обычно не ведут себя в воде как кислоты, но наличие двойной связи, примыкающей к ОН-группе, может существенно уменьшить p K a по механизму кето-енольной таутомерии . Аскорбиновая кислота является примером такого эффекта. Дикетон 2,4-пентандион ( ацетилацетон ) также является слабой кислотой из-за кето-енольного равновесия. В ароматических соединениях, например феноле , имеющих ОН-заместитель, сопряжение с ароматическим кольцом в целом значительно увеличивает стабильность депротонированной формы.

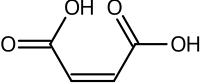
Структурные эффекты также могут иметь важное значение. Разница между фумаровой кислотой и малеиновой кислотой является классическим примером. Фумаровая кислота представляет собой (E)-1,4-бут-2-эндиовую кислоту, транс -изомер , тогда как малеиновая кислота является соответствующим цис- изомером, т.е. (Z)-1,4-бут-2-эндиовой кислотой (см. цис-изомер). транс-изомерия ). Фумаровая кислота имеет значения p K a примерно 3,0 и 4,5. Напротив, малеиновая кислота имеет значения p K a приблизительно 1,5 и 6,5. Причина такой большой разницы заключается в том, что при удалении одного протона от цис- прочная внутримолекулярная водородная связь изомера (малеиновой кислоты) образуется с оставшейся поблизости карбоксильной группой. Это способствует образованию малеата H + , и он выступает против удаления второго протона из этого вида. В транс- изомере две карбоксильные группы всегда расположены далеко друг от друга, поэтому водородная связь не наблюдается. [ 54 ]
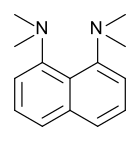
Протонная губка , 1,8-бис(диметиламино)нафталин, имеет значение ap 12,1 K . Это одно из самых сильных известных аминных оснований. Высокая основность объясняется снятием напряжения при протонировании и сильной внутренней водородной связью. [ 55 ] [ 56 ]
В этом разделе также следует упомянуть влияние растворителя и сольватации. Оказывается, эти влияния более тонкие, чем у упомянутой выше диэлектрической среды. Например, ожидаемый (по электронным эффектам метильных заместителей) и наблюдаемый в газовой фазе порядок основности метиламинов Me 3 N > Me 2 NH > MeNH 2 > NH 3 под действием воды меняется на Me 2 NH > MeNH 2 > Me 3 N > NH 3 . Молекулы нейтрального метиламина связываются водородной связью с молекулами воды преимущественно за счет одного акцепторного взаимодействия N–HOH и лишь изредка еще одной донорной связи NH–OH 2 . Следовательно, метиламины стабилизируются гидратацией примерно в одинаковой степени независимо от числа метильных групп. Напротив, соответствующие катионы метиламмония всегда используют все доступные протоны для донорной связи NH–OH 2 . Таким образом, относительная стабилизация ионов метиламмония уменьшается с увеличением числа метильных групп, что объясняет порядок основности воды в метиламинах. [ 4 ]
Термодинамика
[ редактировать ]Константа равновесия связана со стандартным изменением энергии Гиббса для реакции, поэтому для константы диссоциации кислоты
- .

R — газовая постоянная , а T — абсолютная температура . Обратите внимание, что p K a = −log( K a ) и 2,303 ≈ ln (10) . При 25 °C Δ G ⊖ в кДж·моль −1 ≈ 5,708 п Ка моль (1 кДж· −1 = 1000 джоулей на моль ). Свободная энергия состоит из члена энтальпии и члена энтропии . [ 11 ]

Стандартное изменение энтальпии можно определить калориметрически или с помощью уравнения Ван-т-Гоффа , хотя калориметрический метод предпочтительнее. Когда определены как стандартное изменение энтальпии, так и константа диссоциации кислоты, стандартное изменение энтропии легко рассчитать по приведенному выше уравнению. В следующей таблице энтропийные члены рассчитаны на основе экспериментальных значений p K a и Δ H. ⊖ . Данные были тщательно отобраны и относятся к температуре 25 °C и нулевой ионной силе в воде. [ 11 ]
| Сложный | Равновесие | п К а | ΔΔG ⊖ (кДж·моль −1 ) [ д ] | Δ Н ⊖ (кДж·моль −1 ) | − Т Δ С ⊖ (кДж·моль −1 ) [ и ] |
|---|---|---|---|---|---|
| HA = уксусная кислота | ХА ⇌ Ч + + А − | 4.756 | 27.147 | −0.41 | 27.56 |
| H2H2A + = Глицин H + | H2H2A + ⇌ ХА + Ч + | 2.351 | 13.420 | 4.00 | 9.419 |
| ХА ⇌ Ч + + А − | 9.78 | 55.825 | 44.20 | 11.6 | |
| H 2 A = Малеиновая кислота | Ч 2 А ⇌ ХА − + Ч + | 1.92 | 10.76 | 1.10 | 9.85 |
| ХА − ⇌ Ч + + А 2− | 6.27 | 35.79 | −3.60 | 39.4 | |
| H 3 A = Лимонная кислота | Ч 3 А ⇌ Ч 2 А − + Ч + | 3.128 | 17.855 | 4.07 | 13.78 |
| H2H2A − ⇌ ХА 2− + Ч + | 4.76 | 27.176 | 2.23 | 24.9 | |
| ХА 2− ⇌ А 3− + Ч + | 6.40 | 36.509 | −3.38 | 39.9 | |
| H 3 A = Борная кислота | Ч 3 А ⇌ Ч 2 А − + Ч + | 9.237 | 52.725 | 13.80 | 38.92 |
| H 3 A = Фосфорная кислота | Ч 3 А ⇌ Ч 2 А − + Ч + | 2.148 | 12.261 | −8.00 | 20.26 |
| H2H2A − ⇌ ХА 2− + Ч + | 7.20 | 41.087 | 3.60 | 37.5 | |
| ХА 2− ⇌ А 3− + Ч + | 12.35 | 80.49 | 16.00 | 54.49 | |
| ХА − = Сульфат водорода | ХА − ⇌ А 2− + Ч + | 1.99 | 11.36 | −22.40 | 33.74 |
| H 2 A = Щавелевая кислота | Ч 2 А ⇌ ХА − + Ч + | 1.27 | 7.27 | −3.90 | 11.15 |
| ХА − ⇌ А 2− + Ч + | 4.266 | 24.351 | −7.00 | 31.35 |
- ^ Ион водорода как таковой в растворе не существует. Он соединяется с молекулой растворителя; когда растворителем является вода, гидроксония : образуется ион ЧАС + + Н 2 О → Н 3 О + . Эта реакция является количественной и, следовательно, ее можно игнорировать в контексте химического равновесия.
- ^ Обычно принято указывать значения p K , а не K. значения п K знак равно -log 10 K . p K a часто называют константой диссоциации кислоты, но это, строго говоря, неверно, поскольку p K a — колорарифм константы диссоциации.
- ^ В этом определении подразумевается, что частное коэффициентов активности , — константа со значением 1 в заданном наборе экспериментальных условий.
- ^ ΔG ⊖ ≈ 2,303 RT п К а
- ^ Рассчитано здесь на основе значений Δ H и Δ G, указанных в цитате, с использованием − T Δ S. ⊖ = ΔG ⊖ − Δ Н ⊖

| Сложный | Равновесие | п К а | ΔH ⊖ (кДж·моль −1 ) | − Т Δ С ⊖ (кДж·моль −1 ) |
|---|---|---|---|---|
| Б = аммиак | полупансион + ⇌ Б + Ч + | 9.245 | 51.95 | 0.8205 |
| B = Метиламин | полупансион + ⇌ Б + Ч + | 10.645 | 55.34 | 5.422 |
| B = Триэтиламин | полупансион + ⇌ Б + Ч + | 10.72 | 43.13 | 18.06 |
Первое, что следует отметить, это то, что когда p K a положительно, стандартное изменение свободной энергии реакции диссоциации также является положительным. Во-вторых, некоторые реакции экзотермические , а некоторые эндотермические , но когда Δ H ⊖ отрицательно T ΔS ⊖ является доминирующим фактором, определяющим, что Δ G ⊖ является положительным. Наконец, вклад энтропии всегда неблагоприятный ( Δ S ⊖ < 0 ) в этих реакциях. Ионы в водном растворе стремятся ориентировать окружающие молекулы воды, что упорядочивает раствор и уменьшает энтропию. Вклад иона в энтропию представляет собой частичную молярную энтропию, которая часто бывает отрицательной, особенно для небольших или сильно заряженных ионов. [ 57 ] Ионизация нейтральной кислоты включает образование двух ионов, в результате чего энтропия уменьшается ( Δ S ⊖ <0 ). При второй ионизации той же кислоты теперь имеется три иона и анион имеет заряд, поэтому энтропия снова уменьшается.
Обратите внимание, что стандартное изменение свободной энергии реакции относится к изменениям реагентов в их стандартных состояниях к продуктам в их стандартных состояниях. Изменение свободной энергии в состоянии равновесия равно нулю, поскольку химические потенциалы реагентов и продуктов в состоянии равновесия равны.
Экспериментальное определение
[ редактировать ]
Экспериментальное определение значений p K a обычно проводят методом титрования в среде с высокой ионной силой и при постоянной температуре. [ 58 ] Типичная процедура будет следующей. Раствор соединения в среде подкисляют сильной кислотой до полного протонирования соединения. Затем раствор титруют сильным основанием до удаления всех протонов. В каждой точке титрования pH измеряют с помощью стеклянного электрода и pH-метра . Константы равновесия находятся путем подгонки рассчитанных значений pH к наблюдаемым значениям с использованием метода наименьших квадратов . [ 59 ]
Общий объем добавляемого сильного основания должен быть небольшим по сравнению с исходным объемом раствора титранда, чтобы ионная сила оставалась практически постоянной. Это гарантирует, что p K a останется неизменным во время титрования.
Расчетная кривая титрования щавелевой кислоты показана справа. Щавелевая кислота имеет значения p K a 1,27 и 4,27. Следовательно, буферные области будут сосредоточены примерно при pH 1,3 и pH 4,3. Буферные области несут информацию, необходимую для получения значений p K a, поскольку концентрации кислоты и сопряженного основания изменяются вдоль буферной области.
Между двумя буферными областями имеется конечная точка, или точка эквивалентности , примерно при pH 3. Эта конечная точка не является резкой и типична для дипротонной кислоты, буферные области которой перекрываются на небольшую величину: p K a2 − p K. В этом примере a1 составляет около трех. (Если бы разница в значениях p K составляла около двух или меньше, конечная точка не была бы заметна.) Вторая конечная точка начинается примерно при pH 6,3 и является резкой. Это указывает на то, что все протоны были удалены. В этом случае раствор не является буферным, и pH резко возрастает при добавлении небольшого количества сильного основания. Однако уровень pH не может продолжать расти бесконечно. Новая буферная область начинается примерно при pH 11 (p K w - 3), где самоионизация воды становится важной.
Очень трудно измерить значения pH менее двух в водном растворе с помощью стеклянного электрода , поскольку уравнение Нернста не выполняется при таких низких значениях pH. Для определения значений p K менее примерно 2 или более примерно 11 спектрофотометрически [ 60 ] [ 61 ] или ЯМР [ 62 ] [ 63 ] измерения могут использоваться вместо измерений pH или сочетаться с ними.
Когда стеклянный электрод использовать невозможно, как в случае с неводными растворами, часто применяют спектрофотометрические методы. [ 38 ] Они могут включать измерения оптической плотности или флуоресценции . В обоих случаях предполагается, что измеряемая величина пропорциональна сумме вкладов каждого фотоактивного вида; Предполагается , что при измерении поглощения закон Бера-Ламберта применяется .
Изотермическую титровальную калориметрию (ITC) можно использовать для определения как значения ap K , так и соответствующей стандартной энтальпии диссоциации кислоты. [ 64 ] Программное обеспечение для выполнения расчетов поставляется производителями приборов для простых систем.
Водные растворы с обычной водой нельзя использовать для 1 Измерения H ЯМР, кроме тяжелой воды , D 2 O. Вместо этого следует использовать 13 Однако данные С ЯМР можно использовать с обычной водой и 1 Спектры ЯМР Н можно использовать с неводными средами. Величины, измеренные с помощью ЯМР, представляют собой усредненные по времени химические сдвиги , поскольку протонный обмен происходит быстро в шкале времени ЯМР. Другие химические сдвиги, например, 31 P можно измерить.
Микроконстанты
[ редактировать ]
Для некоторых полипротонных кислот диссоциация (или ассоциация) происходит более чем в одном неэквивалентном сайте. [ 4 ] а наблюдаемая макроскопическая константа равновесия, или макроконстанта, представляет собой комбинацию микроконстант, включающих отдельные виды. Когда один реагент образует два продукта параллельно, макроконстанта представляет собой сумму двух микроконстант: Это справедливо, например, для депротонирования аминокислоты цистеина , которая существует в растворе как нейтральный цвиттер-ион. HS-CH 2 -CH(NH + 3 )-COO − . Две микроконстанты представляют собой депротонирование либо по сере, либо по азоту, а сумма макроконстант здесь представляет собой константу диссоциации кислоты. [ 65 ]



Точно так же такое основание, как спермин, имеет более одного места, где может происходить протонирование. Например, монопротонирование может происходить на концевом −NH 2 группа или на внутреннем −NH- группы. Значения K b для диссоциации спермина, протонированного в том или ином из сайтов, являются примерами микроконстант . Их нельзя определить напрямую с помощью измерений pH, поглощения, флуоресценции или ЯМР; Измеренное значение K b представляет собой сумму значений K для микрореакций.

Тем не менее, место протонирования очень важно для биологической функции, поэтому были разработаны математические методы определения микроконстант. [ 66 ]
Когда два реагента параллельно образуют один продукт, макроконстанта [ 65 ] Например, вышеупомянутое равновесие для спермина можно рассматривать с точки зрения значений K a двух таутомерных сопряженных кислот с макроконстантой. В этом случае Это эквивалентно предыдущему выражению, поскольку пропорционально




Когда реагент подвергается двум последовательным реакциям, макроконстанта объединенной реакции представляет собой произведение микроконстанты двух стадий. Например, упомянутый выше цистеиновый цвиттер-ион может потерять два протона, один от серы и один от азота, а общая макроконстанта потери двух протонов является произведением двух констант диссоциации. [ 65 ] Это также можно записать через логарифмические константы как


Приложения и значение
[ редактировать ]Знание значений p K a важно для количественной оценки систем, имеющих кислотно-основное равновесие в растворе. существует множество приложений В биохимии ; например, значения pKa и белков боковых цепей аминокислот имеют большое значение для активности ферментов и стабильности белков. [ 67 ] белка p K a Значения не всегда могут быть измерены напрямую, но могут быть рассчитаны с использованием теоретических методов. Буферные растворы широко используются для получения растворов с физиологическим pH или близким к нему для изучения биохимических реакций; [ 68 ] разработка этих решений зависит от знания значений p K a их компонентов. Важные буферные растворы включают MOPS , который обеспечивает раствор с pH 7,2, и трицин , который используется в гель-электрофорезе . [ 69 ] [ 70 ] Буферизация является важной частью кислотно-щелочной физиологии, включая кислотно-основной гомеостаз . [ 71 ] и является ключом к пониманию таких расстройств, как кислотно-щелочное расстройство . [ 72 ] [ 73 ] [ 74 ] Изоэлектрическая точка данной молекулы является функцией ее значений p K , поэтому разные молекулы имеют разные изоэлектрические точки. Это позволяет использовать технику, называемую изоэлектрической фокусировкой . [ 75 ] который используется для разделения белков с помощью 2-D электрофореза в полиакриламидном геле .
Буферные растворы также играют ключевую роль в аналитической химии . Их используют всякий раз, когда возникает необходимость зафиксировать pH раствора на определенном значении. По сравнению с водным раствором pH буферного раствора относительно нечувствителен к добавлению небольшого количества сильной кислоты или сильного основания. Буферная емкость [ 76 ] простого буферного раствора является наибольшим при pH = p K a . При кислотно-основной экстракции эффективность экстракции соединения в органическую фазу, например эфир , можно оптимизировать путем регулирования pH водной фазы с использованием соответствующего буфера. При оптимальном pH концентрация электрически нейтральных веществ максимальна; такой вид более растворим в органических растворителях с низкой диэлектрической постоянной , чем в воде. Этот метод используется для очистки слабых кислот и оснований. [ 77 ]
Индикатор pH представляет собой слабую кислоту или слабое основание, меняющую цвет в переходном диапазоне pH, который составляет примерно p K a ± 1. Для создания универсального индикатора необходима смесь индикаторов, соседние значения p K a которых отличаются примерно на два, так что их переходные диапазоны pH просто перекрываются.
В фармакологии ионизация соединения изменяет его физическое поведение и макросвойства, такие как растворимость и липофильность , log p ). Например, ионизация любого соединения увеличит растворимость в воде, но уменьшит липофильность. Это используется при разработке лекарств для увеличения концентрации соединения в крови путем регулирования p K a ионизируемой группы. [ 78 ]
Знание значений p K a важно для понимания координационных комплексов , образующихся при взаимодействии иона металла M м+ , действующий как кислота Льюиса , с лигандом L, действующим как основание Льюиса . Однако лиганд может вступать и в реакции протонирования, поэтому образование комплекса в водном растворе можно символически представить реакцией
![{\displaystyle [{\ce {M(H2O)_{\mathit {n}}}}]^{m+}+{\ce {LH<=>}} \ [{\ce {M(H2O)}} _{n-1}{\ce {L}}]^{(m-1)+}+{\ce {H3O+}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/696ff04eb241a2f6a358b8dd1b9c373ea3a8c91d)
Чтобы определить константу равновесия для этой реакции, в которой лиганд теряет протон, необходимо знать p K a протонированного лиганда. На практике лиганд может быть полипротическим; например ЭДТА 4− может принять четыре протона; в этом случае должны быть известны все значения p K a . Кроме того, ион металла подвержен гидролизу , то есть ведет себя как слабая кислота, поэтому необходимо также знать значения p K для реакций гидролиза. [ 79 ]
Оценка опасности, связанной с кислотой или основанием, может потребовать знания pKa значений . [ 80 ] Например, цианистый водород — очень токсичный газ, поскольку ион цианида ингибирует железосодержащий фермент цитохром с-оксидазу . Цианистый водород представляет собой слабую кислоту в водном растворе с ap K a около 9. В сильнощелочных растворах, скажем, с pH выше 11, из этого следует, что цианид натрия «полностью диссоциирован», поэтому опасность, связанная с газообразным цианистым водородом, значительно снижается. . С другой стороны, кислый раствор очень опасен, поскольку весь цианид находится в кислотной форме. Проглатывание цианида через рот потенциально смертельно, независимо от pH, из-за реакции с цитохром-с-оксидазой.
В науке об окружающей среде кислотно-щелочное равновесие важно для озер. [ 81 ] и реки; [ 82 ] [ 83 ] например, гуминовые кислоты являются важными компонентами природных вод. Другой пример встречается в химической океанографии : [ 84 ] Для количественной оценки растворимости железа(III) в морской воде различной солености используются значения p K a для образования продуктов гидролиза железа(III). Fe(ОН) 2+ , Fe(OH) + 2 и Fe(OH) 3 определяли вместе с продуктом растворимости гидроксида железа . [ 85 ]
Значения для обычных веществ
[ редактировать ]Существует несколько методов определения p K a химического вещества, что приводит к некоторым расхождениям между различными источниками. Хорошо измеренные значения обычно находятся в пределах 0,1 единицы друг от друга. Представленные здесь данные были получены при температуре 25 °C в воде. [ 7 ] [ 86 ] Дополнительные значения можно найти в разделе «Термодинамика» выше. Таблицу р Ка углеродных кислот, измеренную в ДМСО, можно найти на странице карбанионов .
| Химическая | Равновесие | п К а |
|---|---|---|
| BH = Аденин | ЧД + 2 ⇌ ЧЧ + Ч + |
4.17 |
| ЧД ⇌ Б - + Ч + | 9.65 | |
| H 3 A = Мышьяковая кислота | Ч 3 А ⇌ Ч 2 А − + Ч + | 2.22 |
| H2H2A − ⇌ ХА 2− + Ч + | 6.98 | |
| ХА 2− ⇌ А 3− + Ч + | 11.53 | |
| HA = Бензойная кислота | ХА ⇌ Ч + + А − | 4.204 |
| HA = Масляная кислота | ХА ⇌ Ч + + А − | 4.82 |
| H 2 A = Хромовая кислота | Ч 2 А ⇌ ХА − + Ч + | 0.98 |
| ХА − ⇌ А 2− + Ч + | 6.5 | |
| Б = Кодеин | ЧД + ⇌ Б + Ч + | 8.17 |
| ГК = Крезол | ХА ⇌ Ч + + А − | 10.29 |
| HA = Муравьиная кислота | ХА ⇌ Ч + + А − | 3.751 |
| HA = плавиковая кислота | ХА ⇌ Ч + + А − | 3.17 |
| HA = синильная кислота | ХА ⇌ Ч + + А − | 9.21 |
| HA = селеноводород | ХА ⇌ Ч + + А − | 3.89 |
| HA = перекись водорода (90%) | ХА ⇌ Ч + + А − | 11.7 |
| HA = молочная кислота | ХА ⇌ Ч + + А − | 3.86 |
| HA = пропионовая кислота | ХА ⇌ Ч + + А − | 4.87 |
| ГК = Фенол | ХА ⇌ Ч + + А − | 9.99 |
| H 2 A = L -(+)-Аскорбиновая кислота | Ч 2 А ⇌ ХА − + Ч + | 4.17 |
| ХА − ⇌ А 2− + Ч + | 11.57 |
См. также
[ редактировать ]- Ацидоз
- Кислоты в вине : винная , яблочная и лимонная — основные кислоты в вине.
- Алкалоз
- Газы артериальной крови
- Химическое равновесие
- Проводимость (электролитическая)
- Механизм Гротгусса : как протоны передаются между ионами гидроксония и молекулами воды, что объясняет исключительно высокую ионную подвижность протона (анимация).
- Функция кислотности Гаммета : мера кислотности, которая используется для очень концентрированных растворов сильных кислот, включая суперкислоты .
- Ионно-транспортное число
- Закисление океана : растворение углекислого газа в атмосфере влияет на pH морской воды . Реакция зависит от общего количества неорганического углерода и от равновесия растворимости с твердыми карбонатами, такими как известняк и доломит .
- Закон разбавления
- рСО2
- рН
- Диаграмма доминирования : относится к равновесиям с участием полиоксианионов . p K a . Для построения этих диаграмм необходимы значения
- Сродство к протону : мера основности в газовой фазе.
- Константы устойчивости комплексов : образование комплекса часто можно рассматривать как конкуренцию между протоном и ионом металла за лиганд, который является продуктом диссоциации кислоты.
Примечания
[ редактировать ]Ссылки
[ редактировать ]- ^ Уиттен, Кеннет В.; Гейли, Кеннет Д.; Дэвис, Раймонд Э. (1992). Общая химия (4-е изд.). Издательство Колледжа Сондерса. п. 660 . ISBN 0-03-072373-6 .
- ^ Петруччи, Ральф Х.; Харвуд, Уильям С.; Херринг, Ф. Джеффри (2002). Общая химия (8-е изд.). Прентис Холл. стр. 667–8 . ISBN 0-13-014329-4 .
- ^ Перрин Д.Д., Демпси Б., Сержант Э.П. (1981). «Глава 3: Методы прогнозирования p K a ». p K a Прогноз для органических кислот и оснований . (вторичный). Лондон: Чепмен и Холл. стр. 21–26. дои : 10.1007/978-94-009-5883-8 . ISBN 978-0-412-22190-3 .
- ^ Jump up to: а б с Фрачкевич Р. (2013). «In Silico предсказание ионизации». В Ридейке Дж. (ред.). Справочный модуль по химии, молекулярным наукам и химической инженерии . (вторичный). Справочный модуль по химии, молекулярным наукам и химической инженерии [онлайн] . Том. 5. Амстердам, Нидерланды: Эльзевир. дои : 10.1016/B978-0-12-409547-2.02610-X . ISBN 9780124095472 .
- ^ Мисслер, Гэри Л.; Тарр, Дональд А. (1991). Неорганическая химия (2-е изд.). Прентис Холл. ISBN 0-13-465659-8 . Глава 6: Кислотно-основная и донорно-акцепторная химия
- ^ Jump up to: а б Белл, Р.П. (1973). Протон в химии (2-е изд.). Лондон: Чепмен и Холл. ISBN 0-8014-0803-2 . Включает обсуждение многих органических кислот Бренстеда.
- ^ Jump up to: а б с Шрайвер, DF; Аткинс, PW (1999). Неорганическая химия (3-е изд.). Оксфорд: Издательство Оксфордского университета. ISBN 0-19-850331-8 . Глава 5: Кислоты и основания
- ^ Хаускрофт, CE; Шарп, AG (2008). Неорганическая химия (3-е изд.). Прентис Холл. ISBN 978-0-13-175553-6 . Глава 6: Кислоты, основания и ионы в водном растворе
- ^ Хедрик, Дж. М.; Дикен, Э.Г.; Уолтерс, РС; Хаммер, Н.И.; Кристи, РА; Кюи, Дж.; Мышакин Е.М.; Дункан, Массачусетс; Джонсон, Массачусетс; Джордан, К.Д. (2005). «Спектральные характеристики колебаний гидратированных протонов в кластерах воды». Наука . 308 (5729): 1765–69. Бибкод : 2005Sci...308.1765H . дои : 10.1126/science.1113094 . ПМИД 15961665 . S2CID 40852810 .
- ^ Смеховский, М.; Стангрет, Дж. (2006). «Гидратация протонов в водном растворе: инфракрасные исследования спектров HDO с преобразованием Фурье». Дж. Хим. Физ . 125 (20): 204508–204522. Бибкод : 2006JChPh.125t4508S . дои : 10.1063/1.2374891 . ПМИД 17144716 .
- ^ Jump up to: а б с Гольдберг, Р.; Кишор, Н.; Леннен, Р. (2002). «Термодинамические величины для реакций ионизации буферов» (PDF) . Дж. Физ. хим. Ссылка. Данные . 31 (2): 231–370. Бибкод : 2002JPCRD..31..231G . дои : 10.1063/1.1416902 . Архивировано из оригинала (PDF) 6 октября 2008 г.
- ^ Джолли, Уильям Л. (1984). Современная неорганическая химия . МакГроу-Хилл. стр. 198 . ISBN 978-0-07-032760-3 .
- ^ Берджесс, Дж. (1978). Ионы металлов в растворе . Эллис Хорвуд. ISBN 0-85312-027-7 . В разделе 9.1 «Кислотность сольватированных катионов» перечислены многие значения p K a .
- ^ Петруччи, Р.Х.; Харвуд, РС; Сельдь, ФГ (2002). Общая химия (8-е изд.). Прентис Холл. ISBN 0-13-014329-4 . стр.698
- ^ Jump up to: а б Россотти, FJC; Россотти, Х. (1961). Определение констант устойчивости . МакГроу-Хилл. Глава 2: Коэффициенты активности и концентрации, стр. 5-10.
- ^ «Проект: поправки ионной силы для констант устойчивости» . Международный союз теоретической и прикладной химии . Проверено 28 марта 2019 г.
- ^ Россотти, Фрэнсис Дж. К.; Розотти, Хейзел (1961). Определение констант устойчивости и других констант равновесия в растворе . Нью-Йорк: МакГроу-Хилл. стр. 5–10. ISBN 9781013909146 . Архивировано из оригинала 7 февраля 2020 года.
- ^ Аткинс, П.В.; де Паула, Дж. (2006). Физическая химия . Издательство Оксфордского университета. ISBN 0-19-870072-5 . Раздел 7.4: Реакция равновесия на температуру
- ^ Петруччи, Ральф Х.; Харвуд, Уильям С.; Херринг, Ф. Джеффри (2002). Общая химия: принципы и современные приложения (8-е изд.). Прентис Холл. п. 633 . ISBN 0-13-014329-4 .
Вам интересно... Как использование активностей делает константу равновесия безразмерной?
- ^ Шрайвер, DF; Аткинс, PW (1999). Неорганическая химия (3-е изд.). Издательство Оксфордского университета. ISBN 0-19-850331-8 . Разд. 5.1c Сильные и слабые кислоты и основания
- ^ Портерфилд, Уильям В. (1984). Неорганическая химия . Аддисон-Уэсли. п. 260. ИСБН 0-201-05660-7 .
- ^ Jump up to: а б Шрайвер, DF; Аткинс, PW (1999). Неорганическая химия (3-е изд.). Издательство Оксфордского университета. ISBN 0-19-850331-8 . Разд. 5.2 Выравнивание растворителем
- ^ Леванов А.В.; Исайкина, О. Я.; Лунин, В.В. (2017). «Константа диссоциации азотной кислоты». Российский журнал физической химии А . 91 (7): 1221–1228. Бибкод : 2017RJPCA..91.1221L . дои : 10.1134/S0036024417070196 . S2CID 104093297 .
- ^ Трумаль, Александр; Липпинг, Лаури; Кальюранд, Ивари; Коппель, Ильмар А.; Лейто, Иво (2016). «Кислотность сильных кислот в воде и диметилсульфоксиде». Журнал физической химии А. 120 (20): 3663–3669. Бибкод : 2016JPCA..120.3663T . дои : 10.1021/acs.jpca.6b02253 . ПМИД 27115918 . S2CID 29697201 .
- ^ Мехта, Акул (22 октября 2012 г.). «Уравнение Хендерсона – Хассельбалха: вывод p K a и p K b » . ФармаXChange . Проверено 16 ноября 2014 г.
- ^ Значения указаны для 25 °C и ионной силы 0 – Пауэлл, Киптон Дж.; Браун, Пол Л.; Бирн, Роберт Х.; Гайда, Тамаш; Хефтер, Гленн; Сьёберг, Стаффан; Ваннер, Ганс (2005). «Химическое образование экологически значимых тяжелых металлов с неорганическими лигандами. Часть 1: Hg 2+ – кл − , ОЙ − , СО3 2- , ТАК 4 2- , и ПО 4 3- водные системы» . Pure Appl. Chem . 77 (4): 739–800. doi : 10.1351/pac200577040739 .
- ^ Браун, TE; Лемей, HE; Берстен, Бельгия; Мерфи, К.; Вудворд, П. (2008). Химия: Центральная наука (11-е изд.). Нью-Йорк: Прентис-Холл. п. 689. ИСБН 978-0-13-600617-6 .
- ^ Jump up to: а б Гринвуд, штат Нью-Йорк; Эрншоу, А. (1997). Химия элементов (2-е изд.). Оксфорд: Баттерворт-Хайнеманн. п. 50. ISBN 0-7506-3365-4 .
- ^ Jump up to: а б с Мисслер, Гэри Л.; Тарр Дональд А. (1999). Неорганическая химия (2-е изд.). Прентис Холл. п. 164. ИСБН 0-13-465659-8 .
- ^ Jump up to: а б Хахи, Джеймс Э. (1983). Неорганическая химия (3-е изд.). Харпер и Роу. п. 297. ИСБН 0-06-042987-9 .
- ^ Лиде, ДР (2004). Справочник CRC по химии и физике, студенческое издание (84-е изд.). ЦРК Пресс. ISBN 0-8493-0597-7 . Секция Д–152
- ^ Скуг, Дуглас А.; Уэст, Дональд М.; Холлер, Ф. Джеймс; Крауч, Стэнли Р. (2014). Основы аналитической химии (9-е изд.). Брукс/Коул. п. 212. ИСБН 978-0-495-55828-6 .
- ^ Хаускрофт, CE; Шарп, АГ (2004). Неорганическая химия (2-е изд.). Прентис Холл. п. 163. ИСБН 978-0-13-039913-7 .
- ^ Харнед, HS; Оуэн, BB (1958). Физическая химия электролитических растворов . Нью-Йорк: Reinhold Publishing Corp., стр. 634–649 , 752–754.
- ^ Jump up to: а б с д Лаудон, Г. Марк (2005), Органическая химия (4-е изд.), Нью-Йорк: Oxford University Press, стр. 317–318, ISBN 0-19-511999-1
- ^ Марч, Дж .; Смит, М. (2007). Продвинутая органическая химия (6-е изд.). Нью-Йорк: Джон Уайли и сыновья. ISBN 978-0-471-72091-1 . Глава 8: Кислоты и основания
- ^ Кютт, А.; Мовчун В.; Родима, Т; Дансауэр, Т.; Русанов Е.Б.; Лейто, И.; Кальюранд, И.; Коппель, Дж.; Пиль, В.; Коппель, И.; Овсянников Г.; Тум, Л.; Мисима, М.; Медебьель, М.; Лорк, Э.; Рёшенталер, ГВ.; Коппель, Айова; Коломейцев, А.А. (2008). «Пентакис (трифторметил) фенил, стерически переполненная и электроноакцепторная группа: синтез и кислотность пентакис (трифторметил) бензола, -толуола, -фенола и -анилина». Дж. Орг. Хим . 73 (7): 2607–2620. дои : 10.1021/jo702513w . ПМИД 18324831 .
- ^ Jump up to: а б Кютт, А.; Лейто, И.; Кальюранд, И.; Соовяли, Л.; Власов В.М.; Ягупольский, Л.М.; Коппель, ИА (2006). «Комплексная самосогласованная спектрофотометрическая шкала кислотности нейтральных кислот Бренстеда в ацетонитриле». Дж. Орг. Хим . 71 (7): 2829–2838. дои : 10.1021/jo060031y . ПМИД 16555839 . S2CID 8596886 .
- ^ Кальюранд, И.; Кютт, А.; Соовяли, Л.; Родима, Т.; Мяэметс, В.; Лейто, я; Коппель, ИА (2005). «Расширение самосогласованной спектрофотометрической шкалы основности в ацетонитриле до полного диапазона 28 единиц пКа: унификация различных шкал основности». Дж. Орг. Хим . 70 (3): 1019–1028. дои : 10.1021/jo048252w . ПМИД 15675863 .
- ^ «Таблица pKa Bordwell (кислотность в ДМСО)» . Архивировано из оригинала 9 октября 2008 года . Проверено 2 ноября 2008 г.
- ^ Хаускрофт, CE; Шарп, AG (2008). Неорганическая химия (3-е изд.). Прентис Холл. ISBN 978-0-13-175553-6 . Глава 8: Неводные среды
- ^ Рочестер, Швейцария (1970). Функции кислотности . Академическая пресса. ISBN 0-12-590850-4 .
- ^ Ола, Джорджия; Пракаш, С; Соммер, Дж (1985). Суперкислоты . Нью-Йорк: Wiley Interscience. ISBN 0-471-88469-3 .
- ^ Кутзи, Дж. Ф.; Падманабхан, Г.Р. (1965). «Сила акцептора протонов и гомоконъюгация моно- и диаминов». Дж. Ам. хим. Соц . 87 (22): 5005–5010. дои : 10.1021/ja00950a006 .
- ^ Сосна, Ш.; Хендриксон, Дж.Б.; Крам, диджей; Хаммонд, GS (1980). Органическая химия . МакГроу-Хилл. п. 203. ИСБН 0-07-050115-7 .
- ^ Бокс, КДж; Вёльги, Г.; Руис, Р.; Комер, Дж. Э.; Такач-Новак, К.; Бош, Э.; Рафолс, К.; Розес, М. (2007). «Физико-химические свойства новой многокомпонентной системы сорастворителей для определения pKa плохо растворимых фармацевтических соединений». Хелв. Хим. Акта . 90 (8): 1538–1553. дои : 10.1002/hlca.200790161 .
- ^ Jump up to: а б Хаускрофт, Кэтрин Э.; Шарп, Алан Г. (2005). Неорганическая химия (2-е изд.). Харлоу, Великобритания: Пирсон Прентис Холл. стр. 170–171. ISBN 0-13-039913-2 .
- ^ Jump up to: а б Дуглас Б., МакДэниел Д.Х. и Александр Дж.Дж. Концепции и модели неорганической химии (2-е изд. Wiley, 1983), стр.526 ISBN 0-471-21984-3
- ^ Полинг, Л. (1960). Природа химической связи и строение молекул и кристаллов; введение в современную структурную химию (3-е изд.). Итака (Нью-Йорк): Издательство Корнельского университета. п. 277 . ISBN 0-8014-0333-2 .
- ^ Сосна, Ш.; Хендриксон, Дж.Б.; Крам, диджей; Хаммонд, GS (1980). Органическая химия . МакГроу-Хилл. ISBN 0-07-050115-7 . Раздел 13-3: Количественные корреляции эффектов заместителей (Часть B) – Уравнение Хэммета
- ^ Хэммет, LP (1937). «Влияние структуры на реакции органических соединений. Производные бензола». Дж. Ам. хим. Соц . 59 (1): 96–103. дои : 10.1021/ja01280a022 .
- ^ Ханш, К.; Лео, А.; Тафт, Р.В. (1991). «Обзор констант заместителей Гаммета, а также параметров резонанса и поля». хим. Преподобный . 91 (2): 165–195. дои : 10.1021/cr00002a004 . S2CID 97583278 .
- ^ Шортер, Дж (1997). «Сборник и критическая оценка параметров и уравнений структурно-реактивности: Часть 2. Расширение шкалы Гаммета σ за счет данных по ионизации замещенных бензойных кислот в водных растворителях при 25 ° C (Технический отчет)» . Чистая и прикладная химия . 69 (12): 2497–2510. дои : 10.1351/pac199769122497 . S2CID 98814841 .
- ^ Сосна, Ш.; Хендриксон, Дж.Б.; Крам, диджей; Хаммонд, GS (1980). Органическая химия . МакГроу-Хилл. ISBN 0-07-050115-7 . Раздел 6-2: Структурное влияние на кислотность и щелочность
- ^ Олдер, RW; Боуман, PS; Стил, WRS; Винтерман, ДР (1968). «Удивительная основность 1,8-бис(диметиламино)нафталина». хим. Коммун. (13): 723–724. дои : 10.1039/C19680000723 .
- ^ Олдер, RW (1989). «Влияние деформации на основность амина». хим. Преподобный . 89 (5): 1215–1223. дои : 10.1021/cr00095a015 .
- ^ Аткинс, Питер Уильям; Де Паула, Хулио (2006). Физическая химия Аткинса . Нью-Йорк: WH Freeman. п. 94 . ISBN 978-0-7167-7433-4 .
- ^ Мартелл, А.Е.; Мотекайтис, Р.Дж. (1992). Определение и использование констант устойчивости . Уайли. ISBN 0-471-18817-4 . Глава 4: Экспериментальная методика потенциометрического измерения pH металлокомплексных равновесий
- ^ Леггетт, диджей (1985). Вычислительные методы определения констант образования . Пленум. ISBN 0-306-41957-2 .
- ^ Аллен, Род-Айленд; Бокс, КДж; Комер, JEA; Пик, К.; Тэм, Кентукки (1998). «Многоволновое спектрофотометрическое определение констант диссоциации кислот ионизируемых лекарственных средств». Дж. Фарм. Биомед. Анал . 17 (4–5): 699–712. дои : 10.1016/S0731-7085(98)00010-7 . ПМИД 9682153 .
- ^ Бокс, КДж; Донкор, Р.Э.; Джапп, Пенсильвания; Лидер, ИП; Трю, Д.Ф.; Тернер, CH (2008). «Химия мультипротонных препаратов. Часть 1: Потенциометрическое многоволновое УФ-титриметрическое исследование и ЯМР-рН-титриметрическое исследование микровидов SKI-606». Дж. Фарм. Биомед. Анал . 47 (2): 303–311. дои : 10.1016/j.jpba.2008.01.015 . ПМИД 18314291 .
- ^ Попов, К.; Ронккомаки, Х.; Лаюнен, LHJ (2006). «Руководство по измерениям ЯМР для определения высоких и низких значений pKa » ( PDF) . Чистое приложение. Хим . 78 (3): 663–675. дои : 10.1351/pac200678030663 . S2CID 4823180 .
- ^ Сакач, З.; Хегеле, Г. (2004). «Точное определение низких p K с помощью значений 1 H ЯМР титрование». Talanta . 62 (4): 819–825. doi : 10.1016/j.talanta.2003.10.007 . PMID 18969368 .
- ^ Фиг, Эндрю Л., изд. (2016). «Методы энзимологии». Калориметрия . 567 . Эльзевир: 2–493. ISSN 0076-6879 .
- ^ Jump up to: а б с Сплиттгербер, АГ; Чинандер, LL (1 февраля 1988 г.). «Спектр промежуточного продукта диссоциации цистеина: биофизический химический эксперимент». Журнал химического образования . 65 (2): 167. Бибкод : 1988JChEd..65..167S . дои : 10.1021/ed065p167 .
- ^ Фрассинети, К.; Альдериги, Л; Ганс, П; Сабатини, А; Вакка, А; Гелли, С. (2003). «Определение констант протонирования некоторых фторированных полиаминов методом 13 Данные ЯМР С, обработанные с помощью новой компьютерной программы HypNMR2000. Последовательность протонирования в полиаминах». Anal. Bioanal. Chem . 376 (7): 1041–1052. : 10.1007 /s00216-003-2020-0 . PMID 12845401. . S2CID 14533024 doi
- ^ Онуфриев А.; Кейс, Д.А.; Ульманн ГМ (2001). «Новый взгляд на титрование pH в биомолекулах». Биохимия . 40 (12): 3413–3419. дои : 10.1021/bi002740q . ПМИД 11297406 .
- ^ Хорошо, НЭ; Вингет, Джорджия; Зима, В.; Коннолли, Теннесси; Идзава, С.; Сингх, РММ (1966). «Буферы ионов водорода для биологических исследований». Биохимия . 5 (2): 467–477. дои : 10.1021/bi00866a011 . ПМИД 5942950 .
- ^ Данн, MJ (1993). Гель-электрофорез: белки . Научные издательства Биос. ISBN 1-872748-21-Х .
- ^ Мартин, Р. (1996). Гель-электрофорез: нуклеиновые кислоты . Научные издательства Биос. ISBN 1-872748-28-7 .
- ^ Бреннер, Б.М.; Штейн, Дж. Х., ред. (1979). Кислотно-щелочной и калиевый гомеостаз . Черчилль Ливингстон. ISBN 0-443-08017-8 .
- ^ Скорпион, Р. (2000). Основы кислот, оснований, буферов и их применение в биохимических системах . Кендалл/Хант Паб. компании ISBN 0-7872-7374-0 .
- ^ Бейнон, Р.Дж.; Истерби, Дж. С. (1996). Буферные растворы: основы . Оксфорд: Издательство Оксфордского университета. ISBN 0-19-963442-4 .
- ^ Перрин, Д.Д.; Демпси, Б. (1974). Буферы для контроля pH и ионов металлов . Лондон: Чепмен и Холл. ISBN 0-412-11700-2 .
- ^ Гарфин , Д.; Ахуджа, С., ред. (2005). Справочник по изоэлектрическому фокусированию и протеомике . Том. 7. Эльзевир. ISBN 0-12-088752-5 .
- ^ Хуланицки, А. (1987). Реакции кислот и оснований в аналитической химии . Массон, MR (редактор перевода). Хорвуд. ISBN 0-85312-330-6 .
- ^ Эяль, AM (1997). «Экстракция кислоты кислотно-основными экстрагентами». Ионный обмен и экстракция растворителями: ряд достижений . 13 : 31–94.
- ^ Авдеев, А. (2003). Абсорбция и разработка лекарств: растворимость, проницаемость и зарядовое состояние . Нью-Йорк: Уайли. ISBN 0-471-42365-3 .
- ^ Бек, Монтана; Надьпал, И. (1990). Химия комплексных равновесий . Хорвуд. ISBN 0-85312-143-5 .
- ^ ван Леувен, CJ; Герменс, Л.М. (1995). Оценка риска химических веществ: Введение . Спрингер. стр. 254–255. ISBN 0-7923-3740-9 .
- ^ Скуг, Д.А.; Уэст, DM; Холлер, Дж. Ф.; Крауч, СР (2004). Основы аналитической химии (8-е изд.). Томсон Брукс/Коул. ISBN 0-03-035523-0 . Глава 9-6: Кислотные дожди и буферная емкость озер
- ^ Штумм, В.; Морган, Джей-Джей (1996). Химия воды . Нью-Йорк: Уайли. ISBN 0-471-05196-9 .
- ^ Снойинк, В.Л.; Дженкинс, Д. (1980). Водная химия: химическое равновесие и скорости в природных водах . Нью-Йорк: Уайли. ISBN 0-471-51185-4 .
- ^ Миллеро, Ф.Дж. (2006). Химическая океанография (3-е изд.). Лондон: Тейлор и Фрэнсис. ISBN 0-8493-2280-4 .
- ^ Миллеро, Ф.Дж.; Лю, X. (2002). «Растворимость железа в морской воде». Морская химия . 77 (1): 43–54. Бибкод : 2002Март..77...43Л . дои : 10.1016/S0304-4203(01)00074-3 .
- ^ Спейт, Дж. Г. (2005). Справочник Ланге по химии (18-е изд.). МакГроу-Хилл. ISBN 0-07-143220-5 . Глава 8
Дальнейшее чтение
[ редактировать ]- Альберт, А.; Сержант, EP (1971). Определение констант ионизации: Лабораторное пособие . Чепмен и Холл. ISBN 0-412-10300-1 . (Предыдущее издание опубликовано как Константы ионизации кислот и оснований . Лондон (Великобритания): Метуэн. 1962. )
- Аткинс, П.В.; Джонс, Л. (2008). Химические принципы: В поисках понимания (4-е изд.). У. Х. Фриман. ISBN 978-1-4292-0965-6 .
- Хаускрофт, CE; Шарп, AG (2008). Неорганическая химия (3-е изд.). Прентис Холл. ISBN 978-0-13-175553-6 . (Неводные растворители)
- Хуланицки, А. (1987). Реакции кислот и оснований в аналитической химии . Хорвуд. ISBN 0-85312-330-6 . (редактор перевода: Мэри Р. Мэссон)
- Перрин, Д.Д.; Демпси, Б.; Сержант, EP (1981). Прогноз рКа для органических кислот и оснований . Чепмен и Холл. ISBN 0-412-22190-Х .
- Райхардт, К. (2003). Растворители и эффекты растворителей в органической химии (3-е изд.). Вайли-ВЧ. ISBN 3-527-30618-8 . Глава 4: Влияние растворителей на положение гомогенного химического равновесия.
- Скуг, Д.А.; Уэст, DM; Холлер, Дж. Ф.; Крауч, СР (2004). Основы аналитической химии (8-е изд.). Томсон Брукс/Коул. ISBN 0-03-035523-0 .
Внешние ссылки
[ редактировать ]- Данные о кислотности и основности в неводных растворителях. Обширная библиография значений p K a в ДМСО , ацетонитриле , ТГФ , гептане , 1,2-дихлорэтане и в газовой фазе.
- Curtipot Бесплатное программное обеспечение «все в одном» для расчета pH и кислотно-щелочного равновесия, а также для моделирования и анализа кривых потенциометрического титрования с помощью электронных таблиц.
- Калькулятор физических/химических свойств SPARC для водных, неводных и газообразных фаз Включает базу данных со значениями p K a , поиск по которым можно осуществлять с помощью SMILES или CAS. регистрационных номеров
- Константы водного равновесия Значения p K a для различных кислот и оснований. Включает таблицу некоторых продуктов растворимости.
- Бесплатное руководство по p K a и log p. интерпретации и измерению Архивировано 10 августа 2016 г. на Wayback Machine. Объяснения значимости этих свойств для фармакологии.
- Бесплатный онлайн-инструмент для прогнозирования (Марвин) p K a , log p , log d и т. д. От ChemAxon
- Chemicalize.org : Список свойств на основе предсказанной структуры.
- p K a Диаграмма [1] Дэвида А. Эванса