Митохондриальная генетика человека
| Митохондриальная ДНК человека | |
|---|---|
 человека длиной 16 569 п.н. Митохондриальный геном с генами, кодирующими белки (красный, оранжевый, желтый), рибосомальной РНК (синий) и генами транспортной РНК (белый). Некодирующая контрольная область мтДНК выделена серым цветом. | |
| Функции | |
| Длина ( б.н. ) | 16,569 |
| Количество генов | 13 (кодирующие гены) 24 ( некодирующие гены ) |
| Тип | Митохондриальная ДНК |
| Полные списки генов | |
| HGNC | Список генов |
| NCBI | Список генов |
| Внешние программы просмотра карт | |
| Вместе | Хромосома МТ |
| Входить | Хромосома МТ |
| NCBI | Хромосома МТ |
| УКСК | Хромосома М |
| Полные последовательности ДНК | |
| RefSeq | NC_012920 ( БЫСТРО ) |
| ГенБанк | J01415 ( БЫСТРО ) |

Митохондриальная генетика человека — это изучение генетики митохондриальной ДНК ( человека ДНК , содержащейся в митохондриях человека ). Митохондриальный геном человека — это совокупность наследственной информации, содержащейся в митохондриях человека. Митохондрии — это небольшие структуры в клетках , которые генерируют энергию для использования клеткой, и поэтому их называют «электростанциями» клетки.
Митохондриальная ДНК (мтДНК) не передается через ядерную ДНК (нДНК). У человека, как и у большинства многоклеточных организмов, митохондриальная ДНК наследуется только от материнской яйцеклетки . Однако существуют теории, что передача мтДНК по отцовской линии у человека может происходить при определенных обстоятельствах. [ 3 ] Митохондриальная наследственность, следовательно, неменделевская , поскольку менделевская наследственность предполагает, что половина генетического материала оплодотворенной яйцеклетки ( зиготы ) происходит от каждого родителя.
Это позволило создать гаплогруппы митохондриальной ДНК для изучения популяционной генетики .
Восемьдесят процентов митохондриальной ДНК кодируют митохондриальную РНК, и поэтому большинство мутаций митохондриальной ДНК приводят к функциональным проблемам, которые могут проявляться в виде мышечных расстройств ( миопатий ).
Поскольку митохондрии обеспечивают 30 молекул АТФ на молекулу глюкозы в отличие от 2 молекул АТФ, образующихся при гликолизе , митохондрии необходимы всем высшим организмам для поддержания жизни. Митохондриальные заболевания — это генетические нарушения, переносимые митохондриальной ДНК или ядерной ДНК, кодирующей митохондриальные компоненты. Небольшие проблемы с любым из многочисленных ферментов, используемых митохондриями, могут иметь разрушительные последствия для клетки и, в свою очередь, для организма.
Количество
[ редактировать ]У человека митохондриальная ДНК (мтДНК) образует замкнутые кольцевые молекулы, содержащие 16 569 [ 4 ] [ 5 ] ДНК пары оснований , [ 6 ] при этом каждая такая молекула обычно содержит полный набор митохондриальных генов. Каждая митохондрия человека содержит в среднем примерно 5 таких молекул мтДНК, количество которых колеблется от 1 до 15. [ 6 ] Каждая клетка человека содержит около 100 митохондрий, что дает общее количество молекул мтДНК на клетку человека около 500. [ 6 ] Количество митохондрий на клетку также варьируется в зависимости от типа клеток, вот некоторые примеры:
- Эритроциты : 0 митохондрий на клетку. [ 1 ]
- Лимфоциты : 3 митохондрии на клетку. [ 7 ]
- Яйцеклетка : Зрелые яйцеклетки метафазы II могут содержать 100 000 митохондрий и 50 000–1 500 000 копий митохондриального генома (что соответствует до 90% ДНК яйцеклетки). [ 2 ]
Шаблоны наследования
[ редактировать ]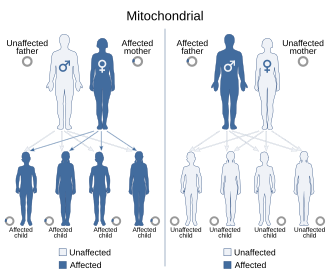

Поскольку митохондриальные заболевания (заболевания, вызванные нарушением работы митохондрий) могут наследоваться как по материнской линии, так и посредством хромосомного наследования, способ их передачи из поколения в поколение может сильно различаться в зависимости от заболевания. Митохондриальные генетические мутации, возникающие в ядерной ДНК, могут возникать в любой из хромосом (в зависимости от вида). Мутации, наследуемые через хромосомы, могут быть аутосомно-доминантными или рецессивными, а также могут быть сцепленными с полом доминантными или рецессивными. Хромосомная наследственность подчиняется обычным менделевским законам , несмотря на то, что фенотип заболевания может быть замаскирован.
Из-за сложных способов «коммуникации» и взаимодействия митохондриальной и ядерной ДНК трудно диагностировать даже, казалось бы, простое наследование. Мутация хромосомной ДНК может изменить белок, который регулирует (увеличивает или уменьшает) продукцию другого определенного белка в митохондриях или цитоплазме; это может привести к легким, если таковые имеются, заметным симптомам. С другой стороны, некоторые разрушительные мутации мтДНК легко диагностировать из-за их широко распространенного повреждения мышечных, нервных и/или печеночных тканей (среди других высокоэнергетических и зависимых от метаболизма тканей), а также потому, что они присутствуют у матери и всех остальных тканей. потомство.
Число затронутых молекул мтДНК, унаследованных конкретным потомком, может сильно различаться, поскольку
- митохондрии внутри оплодотворенного яйцеклетки — это то, с чего должна начаться новая жизнь (с точки зрения мтДНК),
- количество пораженных митохондрий варьируется от клетки (в данном случае оплодотворенного ооцита) к клетке в зависимости как от количества, унаследованного от материнской клетки, так и от факторов окружающей среды, которые могут благоприятствовать мутантной или дикой типа митохондриальной ДНК,
- количество молекул мтДНК в митохондриях варьируется от двух до десяти.
Даже при рождении близнецов возможно, что один ребенок получит более половины мутантных молекул мтДНК, в то время как другой близнец может получить лишь небольшую часть мутантных молекул мтДНК по отношению к дикому типу (в зависимости от того, как близнецы делятся друг от друга и как по обе стороны деления оказывается множество мутантных митохондрий). В некоторых случаях некоторое количество митохондрий или митохондрии из сперматозоида попадают в ооцит, но отцовские митохондрии активно разлагаются.
Гены
[ редактировать ]Гены в митохондриальном геноме человека следующие.
Электронно-транспортная цепь и гуманин
[ редактировать ]Первоначально ошибочно считалось, что митохондриальный геном содержит только 13 белок-кодирующих генов, причем все они кодируют белки цепи переноса электронов . Однако в 2001 году был открыт 14-й биологически активный белок, названный гуманином , который кодируется митохондриальным геном MT-RNR2, который также кодирует часть митохондриальной рибосомы (состоящей из РНК):
| Сложный число |
Категория | Гены | Позиции в митогеноме | Стрэнд |
|---|---|---|---|---|
| я | НАДН-дегидрогеназа | |||
| МТ-НД1 | 3,307–4,262 | ЧАС | ||
| МТ-НД2 | 4,470–5,511 | ЧАС | ||
| МТ-НД3 | 10,059–10,404 | ЧАС | ||
| МТ-НД4Л | 10,470–10,766 | ЧАС | ||
| МТ-НД4 | 10 760–12 137 (перекрытие с МТ-НД4Л) | ЧАС | ||
| МТ-НД5 | 12,337–14,148 | ЧАС | ||
| МТ-НД6 | 14,149–14,673 | л | ||
| III | Коэнзим Q - цитохром с редуктаза / цитохром b | МТ-КИБ | 14,747–15,887 | ЧАС |
| IV | Цитохром с оксидаза | МТ-СО1 | 5,904–7,445 | ЧАС |
| МТ-CO2 | 7,586–8,269 | ЧАС | ||
| МТ-CO3 | 9,207–9,990 | ЧАС | ||
| V | АТФ-синтаза | МТ-АТФ6 | 8527–9207 (перекрытие с MT-ATP8) | ЧАС |
| МТ-АТП8 | 8,366–8,572 | ЧАС | ||
| — | Человек | МТ-РНР2 | — | — |
В отличие от других белков, гуманин не остается в митохондриях, а взаимодействует с остальной частью клетки и клеточными рецепторами. Гуманин может защищать клетки мозга, ингибируя апоптоз . Несмотря на название, версии гуманина существуют и у других животных, например, у крыс.
рРНК
[ редактировать ]Следующие гены кодируют рРНК :
| Субъединица | рРНК | Гены | Позиции в митогеноме | Стрэнд |
|---|---|---|---|---|
| Малый (СГУ) | 12С | МТ-РНР1 | 648–1,601 | ЧАС |
| Большой (ЛСУ) | 16С | МТ-РНР2 | 1,671–3,229 | ЧАС |
тРНК
[ редактировать ]Следующие гены кодируют тРНК :
| Аминокислота | 3-буквенный | 1-буква | МТ ДНК | Позиции | Стрэнд |
|---|---|---|---|---|---|
| Аланин | Земля | А | МТ-ТА | 5,587–5,655 | л |
| Аргинин | Арг | Р | MT-TR | 10,405–10,469 | ЧАС |
| Аспарагин | Асн | Н | МТ-ТН | 5,657–5,729 | л |
| Аспарагиновая кислота | Асп | Д | МТ-ТД | 7,518–7,585 | ЧАС |
| Цистеин | Цис | С | МТ-ТС | 5,761–5,826 | л |
| Глутаминовая кислота | Глу | И | МТ-ТЭ | 14,674–14,742 | л |
| Глютамин | Глн | вопрос | МТ-ТК | 4,329–4,400 | л |
| Глицин | Глай | Г | МТ-ТГ | 9,991–10,058 | ЧАС |
| Гистидин | Его | ЧАС | МТ-ТД | 12,138–12,206 | ЧАС |
| изолейцин | С | я | МТ-ТИ | 4,263–4,331 | ЧАС |
| Лейцин | Лей (ЧАС) | л | МТ-ТЛ1 | 3,230–3,304 | ЧАС |
| Лейцин | Лео (КУН) | л | МТ-ТЛ2 | 12,266–12,336 | ЧАС |
| Лизин | Свет | К | МТ-ТК | 8,295–8,364 | ЧАС |
| Метионин | Из | М | МТ-ТМ | 4,402–4,469 | ЧАС |
| Фенилаланин | Пхе | Ф | МТ-ТФ | 577–647 | ЧАС |
| Пролин | Про | П | МТ-ТП | 15,956–16,023 | л |
| Серин | Бытие (UCN) | С | МТ-ТС1 | 7,446–7,514 | л |
| Серин | Сыр (МОЗГ) | С | МТ-ТС2 | 12,207–12,265 | ЧАС |
| Треонин | чр | Т | МТ-ТТ | 15,888–15,953 | ЧАС |
| Триптофан | Трп | В | МТ-ТВ | 5,512–5,579 | ЧАС |
| Тирозин | Тир | И | MT-TY | 5,826–5,891 | л |
| Валин | Вал | V | МТ-ТВ | 1,602–1,670 | ЧАС |
Расположение генов
[ редактировать ]Митохондриальная ДНК традиционно имела две цепи ДНК, обозначаемые как тяжелая и легкая цепь, из-за их плавучей плотности во время разделения в градиентах хлорида цезия. [ 8 ] [ 9 ] Было обнаружено, что это связано с относительным содержанием нуклеотидов G+T в цепи. [ 10 ] Однако путаница в маркировке этих цепей широко распространена и, по-видимому, возникла из-за того, что в одной влиятельной статье 1999 года большая часть кодирующей цепи была идентифицирована как тяжелая. [ 11 ] [ 10 ] У человека легкая цепь мтДНК несет 28 генов, а тяжелая цепь мтДНК несет только 9 генов. [ 10 ] [ 12 ] Восемь из девяти генов тяжелой цепи кодируют молекулы митохондриальной тРНК. МтДНК человека состоит из 16 569 пар нуклеотидов. Вся молекула регулируется только одной регуляторной областью, которая содержит точки начала репликации как тяжелых, так и легких цепей. Полная молекула митохондриальной ДНК человека картирована [1] [2] .
Варианты генетического кода
[ редактировать ]Генетический код по большей части универсален, за некоторыми исключениями: [ 13 ] митохондриальная генетика включает некоторые из них. Для большинства организмов « стоп-кодонами » являются «UAA», «UAG» и «UGA». В митохондриях позвоночных «AGA» и «AGG» также являются стоп-кодонами, но не «UGA», который вместо этого кодирует триптофан . «AUA» кодирует изолейцин у большинства организмов, но метионин в мРНК митохондрий позвоночных.
Существует множество других вариаций кодов, используемых другими митохондриальными м/тРНК, которые не оказались вредными для их организмов и которые можно использовать в качестве инструмента (наряду с другими мутациями среди мтДНК/РНК разных видов) для определения относительная близость общего происхождения родственных видов. (Чем более родственными являются два вида, тем больше мутаций мтДНК/РНК будет одинаковыми в их митохондриальном геноме).
Используя эти методы, было подсчитано, что первые митохондрии возникли около 1,5 миллиардов лет назад. Общепринятая гипотеза состоит в том, что митохондрии возникли из аэробных прокариот в симбиотических отношениях с анаэробными эукариотами .
Репликация, репарация, транскрипция и трансляция
[ редактировать ]Репликация митохондрий контролируется ядерными генами и специально приспособлена для создания такого количества митохондрий, которое необходимо конкретной клетке в данный момент.
Транскрипция митохондрий у человека инициируется тремя промоторами : H1, H2 и L (промоторы тяжелой цепи 1, тяжелой цепи 2 и промоторы легкой цепи). Промотор H2 транскрибирует почти всю тяжелую цепь, а промотор L — всю легкую цепь. Промотор H1 вызывает транскрипцию двух митохондриальных молекул рРНК. [ 14 ]
Когда транскрипция происходит на тяжелой цепи, создается полицистронный транскрипт. Легкая цепь производит либо небольшие транскрипты, которые можно использовать в качестве праймеров , либо один длинный транскрипт. Производство праймеров происходит путем обработки транскриптов легкой цепи с помощью митохондриальной РНКазы MRP (процессинг митохондриальной РНК). Необходимость транскрипции для производства праймеров связывает процесс транскрипции с репликацией мтДНК. Полноразмерные транскрипты разрезаются на функциональные молекулы тРНК, рРНК и мРНК. [ нужна ссылка ]
В процессе инициации транскрипции в митохондриях участвуют три типа белков: митохондриальная РНК-полимераза ( POLRMT ), митохондриальный фактор транскрипции А (TFAM) и митохондриальные факторы транскрипции B1 и B2 (TFB1M, TFB2M). POLRMT , TFAM и TFB1M или TFB2M собираются на митохондриальных промоторах и начинают транскрипцию. Фактические молекулярные события, участвующие в инициации, неизвестны, но эти факторы составляют базальный механизм транскрипции и, как было показано, функционируют in vitro. [ нужна ссылка ]
Митохондриальный перевод до сих пор не очень хорошо изучен. Трансляции in vitro до сих пор не увенчались успехом, вероятно, из-за сложности выделения достаточного количества мРНК, функциональной мРНК и, возможно, из-за сложных изменений, которые мРНК претерпевает перед трансляцией. [ нужна ссылка ]
Митохондриальная ДНК-полимераза
[ редактировать ]Митохондриальная ДНК-полимераза (Pol gamma, кодируемая геном POLG ) используется для копирования мтДНК во время репликации. Поскольку две цепи ( тяжелая и легкая ) кольцевой молекулы мтДНК имеют разные начала репликации , она реплицируется в режиме D-петли . Одна цепь начинает реплицироваться первой, вытесняя другую. Это продолжается до тех пор, пока репликация не достигнет начала репликации на другой цепи, после чего другая цепь начинает реплицироваться в противоположном направлении. В результате появляются две новые молекулы мтДНК. Каждая митохондрия имеет несколько копий молекулы мтДНК, и количество молекул мтДНК является ограничивающим фактором деления митохондрий . После того, как митохондрия наберет достаточно мтДНК, площади мембраны и мембранных белков, она может подвергнуться делению (очень похожему на то, которое используют бактерии), чтобы стать двумя митохондриями. Имеющиеся данные свидетельствуют о том, что митохондрии также могут подвергаться слиянию и обмену (в форме кроссовера ) генетическим материалом между собой. Митохондрии иногда образуют большие матрицы, в которых Слияние , деление и белковый обмен происходят постоянно. мтДНК является общей для митохондрий (несмотря на то, что они могут подвергаться слиянию). [ нужна ссылка ]
Повреждение и ошибка транскрипции
[ редактировать ]Митохондриальная ДНК подвержена повреждению свободными радикалами кислорода из-за ошибок, которые происходят во время производства АТФ через цепь переноса электронов. Эти ошибки могут быть вызваны генетическими нарушениями, раком и перепадами температуры. Эти радикалы могут повредить молекулы мтДНК или изменить их, затрудняя их репликацию митохондриальной полимеразой. Оба случая могут привести к делециям, перестройкам и другим мутациям. Недавние данные свидетельствуют о том, что в митохондриях есть ферменты, которые считывают мтДНК и исправляют мутации, которые могут возникнуть из-за свободных радикалов. Считается, что ДНК-рекомбиназа, обнаруженная в клетках млекопитающих, также участвует в процессе репарационной рекомбинации. Делеции и мутации, вызванные свободными радикалами, связаны с процессом старения. Считается, что радикалы вызывают мутации, которые приводят к появлению мутантных белков, что, в свою очередь, приводит к увеличению количества радикалов. Этот процесс занимает много лет и связан с некоторыми процессами старения, затрагивающими кислородзависимые ткани, такие как мозг, сердце, мышцы и почки. Подобные процессы самоусиления являются возможными причинами дегенеративных заболеваний, в том числе Болезнь Паркинсона , Альцгеймера и ишемическая болезнь сердца . [ нужна ссылка ]
Хромосомно-опосредованные ошибки репликации мтДНК
[ редактировать ]Поскольку рост и деление митохондрий опосредованы ядерной ДНК, мутации в ядерной ДНК могут иметь широкий спектр эффектов на репликацию мтДНК. Несмотря на то, что локусы некоторых из этих мутаций были обнаружены в хромосомах человека, конкретные гены и белки, участвующие в этом процессе, еще не выделены. Для деления митохондриям необходим определенный белок. Если этого белка (генерируемого ядром) нет, митохондрии растут, но не делятся. Это приводит к образованию гигантских, неэффективных митохондрий. Ошибки в хромосомных генах или их продуктах также могут более непосредственно влиять на репликацию митохондрий, ингибируя митохондриальную полимеразу, и даже могут прямо или косвенно вызывать мутации в мтДНК. Косвенные мутации чаще всего вызываются радикалами, созданными дефектными белками, состоящими из ядерной ДНК. [ нужна ссылка ]
Митохондриальные заболевания
[ редактировать ]Вклад митохондриального и ядерного генома
[ редактировать ]Всего в митохондриях содержится около 3000 различных типов белков, но только около 13 из них кодируются митохондриальной ДНК. Большинство из 3000 типов белков участвуют в различных процессах, помимо производства АТФ, таких как синтез порфиринов . Лишь около 3% из них кодируют белки, вырабатывающие АТФ. Это означает, что большая часть генетической информации, кодирующей белковый состав митохондрий, находится в хромосомной ДНК и участвует в других процессах, помимо синтеза АТФ. Это увеличивает вероятность того, что мутация, которая затронет митохондрии, произойдет в хромосомной ДНК, которая наследуется по менделевскому типу. Другой результат заключается в том, что хромосомная мутация повлияет на конкретную ткань из-за ее особых потребностей, будь то высокие энергетические потребности или потребность в катаболизме или анаболизме определенного нейромедиатора или нуклеиновой кислоты. Поскольку каждая митохондрия несет несколько копий митохондриального генома (2–10 у человека), митохондриальные мутации могут наследоваться по материнской линии за счет мутаций мтДНК, которые присутствуют в митохондриях внутри клетки. яйцеклетки до оплодотворения или (как указано выше) посредством мутаций в хромосомах. [ нужна ссылка ]
Презентация
[ редактировать ]Митохондриальные заболевания варьируются по степени тяжести от бессимптомных до смертельных и чаще всего возникают из-за наследственных, а не приобретенных мутаций митохондриальной ДНК. Данная митохондриальная мутация может вызывать различные заболевания в зависимости от серьезности проблемы в митохондриях и ткани, в которой находятся пораженные митохондрии. И наоборот, несколько различных мутаций могут проявляться как одно и то же заболевание. Из-за такой почти индивидуальной характеристики митохондриальных заболеваний (см. Персонализированная медицина ) их очень сложно точно распознать, диагностировать и отследить. Некоторые заболевания наблюдаются при рождении или даже до него (многие из них приводят к смерти), тогда как другие не проявляются до позднего взросления (расстройства с поздним началом). Это связано с тем, что количество митохондрий мутантного и дикого типа варьируется в зависимости от клеток и тканей и постоянно меняется. Поскольку клетки имеют несколько митохондрий, разные митохондрии в одной и той же клетке могут иметь разные варианты мтДНК . Это состояние называется гетероплазмия . Когда в определенной ткани достигается определенное соотношение митохондрий мутантного и дикого типа, возникает заболевание. Соотношение варьируется от человека к человеку и от ткани к ткани (в зависимости от ее конкретных потребностей в энергии, кислороде и метаболизме, а также эффектов конкретной мутации). Митохондриальные заболевания очень многочисленны и разнообразны. Помимо заболеваний, вызванных аномалиями митохондриальной ДНК, предполагается, что многие заболевания частично связаны с митохондриальными дисфункциями, такими как сахарный диабет , [ 15 ] формы рака [ 16 ] и сердечно-сосудистые заболевания , лактоацидоз , [ 17 ] специфические формы миопатии , [ 18 ] остеопороз , [ 19 ] болезнь Альцгеймера , [ 20 ] болезнь Паркинсона , [ 21 ] гладить , [ 22 ] мужское бесплодие [ 23 ] и которые, как также полагают, играют роль в процессе старения . [ 24 ]
Использование в криминалистике
[ редактировать ]МтДНК человека также можно использовать для идентификации людей. [ 25 ] Судебно-медицинские лаборатории иногда используют сравнение мтДНК для идентификации человеческих останков, особенно для идентификации старых неопознанных останков скелетов. Хотя в отличие от ядерной ДНК мтДНК не является специфичной для одного человека, ее можно использовать в сочетании с другими доказательствами (антропологическими данными, косвенными доказательствами и т.п.) для установления идентификации. мтДНК также используется для исключения возможных совпадений между пропавшими без вести людьми и неопознанными останками. [ 26 ] Многие исследователи считают, что мтДНК лучше подходит для идентификации более старых останков скелета, чем ядерная ДНК, поскольку большее количество копий мтДНК на клетку увеличивает вероятность получения полезного образца, а также потому, что совпадение с живым родственником возможно, даже если многочисленные материнские поколения разделяют эти два понятия.
Примеры
[ редактировать ]Останки американского преступника Джесси Джеймса были идентифицированы путем сравнения мтДНК, извлеченной из его останков, и мтДНК сына правнучки его сестры по женской линии. [ 27 ]
Точно так же останки Александры Федоровны (Аликс Гессенской) , последней императрицы России, и ее детей были идентифицированы путем сравнения их митохондриальной ДНК с ДНК принца Филиппа, герцога Эдинбургского , чья бабушка по материнской линии была сестрой Александры Викторией Гессенской . [ 28 ]
Аналогичным образом, для идентификации останков императора Николая II его митохондриальную ДНК сравнили с ДНК Джеймса Карнеги, 3-го герцога Файфского , чья прабабушка по материнской линии Александра Датская (королева Александра) была сестрой матери Николая II Дагмар Датской (императрица Мария Федоровна). [ 28 ] [ 29 ]
Аналогичным образом были идентифицированы останки короля Ричарда III . [ 30 ]
См. также
[ редактировать ]- Передача мтДНК по отцовской линии
- Гаплогруппы митохондриальной ДНК человека
- Кембриджская эталонная последовательность
- Митохондриальные молекулярные часы человека
- Генетическая генеалогия для списков баз данных, которые помогают пользователям находить других людей по их Y-ДНК и мтДНК.
Ссылки
[ редактировать ]- ^ Перейти обратно: а б Шустер Р.К., Рубинштейн А.Дж., Уоллес округ Колумбия (1988). «Митохондриальная ДНК в безъядерных клетках крови человека» . Биохимия Биофиз Рес Коммьюнити . 155 (3): 1360–5. дои : 10.1016/s0006-291x(88)81291-9 . ПМИД 3178814 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Перейти обратно: а б Чжан Д., Кейлти Д., Чжан З.Ф., Чиан Р.К. (2017). «Митохондрии в старении ооцитов: современное понимание» . Факты Мнения Вис Обгын . 9 (1): 29–38. ПМК 5506767 . ПМИД 28721182 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Шварц, Марианна; Виссинг, Джон (22 августа 2002 г.). «Отцовское наследование митохондриальной ДНК» . Медицинский журнал Новой Англии . 347 (8): 576–580. дои : 10.1056/NEJMoa020350 . ПМИД 12192017 .
- ^ Андерсон, С.; Банкир, AT; Баррелл, Б.Г.; де Брёйн, МХЛ; Коулсон, Арканзас; Друэн, Дж.; Эперон, ИЦ; Нирлих, ДП; Роу, бакалавр; Сэнгер, Ф.; Шрайер, PH; Смит, AJH; Стаден, Р.; Янг, И.Г. (апрель 1981 г.). «Последовательность и организация митохондриального генома человека». Природа . 290 (5806): 457–465. Бибкод : 1981Natur.290..457A . дои : 10.1038/290457a0 . ПМИД 7219534 . S2CID 4355527 .
- ^ «Без названия» . Архивировано из оригинала 13 августа 2011 г. Проверено 13 июня 2012 г.
- ^ Перейти обратно: а б с Сато, М; Куроива, Т. (сентябрь 1991 г.). «Организация множества нуклеоидов и молекул ДНК в митохондриях клетки человека». Экспериментальные исследования клеток . 196 (1): 137–140. дои : 10.1016/0014-4827(91)90467-9 . ПМИД 1715276 .
- ^ Надь Г., Барча М., Гончоров Н., Филлипс П.Е., Перл А. (2004). «Зависимый от оксида азота митохондриальный биогенез генерирует сигнальный профиль Ca2+ Т-клеток волчанки» . Дж Иммунол . 173 (6): 3676–83. дои : 10.4049/jimmunol.173.6.3676 . ПМК 4034140 . ПМИД 15356113 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Циммерман, граф Г.; Акинс, Дэррин Р.; Планц, Джон В.; Шурр, Майкл Дж. (сентябрь 1988 г.). «Быстрая процедура выделения митохондриальной ДНК». Методы генного анализа . 5 (5): 102–104. дои : 10.1016/0735-0651(88)90004-0 . ПМИД 2847966 .
- ^ Велтер, Корнелиус; Миз, Эккарт; Блин, Николаус (1988). «Быстрая ступенчатая градиентная очистка митохондриальной ДНК». Отчеты по молекулярной биологии . 13 (2): 117–120. дои : 10.1007/BF00539059 . ПМИД 3221842 . S2CID 3157709 .
- ^ Перейти обратно: а б с Баррозу Лима, Николас Коста; Просдочими, Франциско (17 февраля 2018 г.). «Дилемма тяжелой цепи митохондрий позвоночных в зависимости от возраста секвенирования генома: количество кодируемых генов или содержание G + T?». Митохондриальная ДНК Часть А. 29 (2): 300–302. дои : 10.1080/24701394.2016.1275603 . ПМИД 28129726 . S2CID 20552678 .
- ^ Таанман, Ян-Виллем (февраль 1999 г.). «Митохондриальный геном: структура, транскрипция, трансляция и репликация» . Biochimica et Biophysica Acta (BBA) — Биоэнергетика . 1410 (2): 103–123. дои : 10.1016/s0005-2728(98)00161-3 . ПМИД 10076021 .
- ^ Андерсон, С.; Банкир, AT; Баррелл, Б.Г.; де Брёйн, МХЛ; Коулсон, Арканзас; Друэн, Дж.; Эперон, ИЦ; Нирлих, ДП; Роу, бакалавр; Сэнгер, Ф.; Шрайер, PH; Смит, AJH; Стаден, Р.; Янг, И.Г. (1981). «Последовательность и организация митохондриального генома человека». Природа . 290 (5806): 457–65. Бибкод : 1981Natur.290..457A . дои : 10.1038/290457a0 . ПМИД 7219534 . S2CID 4355527 .
- ^ «Генетические коды» . www.ncbi.nlm.nih.gov . Национальный центр биотехнологической информации . Проверено 16 марта 2019 г.
- ^ Асин-Каюэла, Хорди; Густафссон, Клаас М. (2007). «Митохондриальная транскрипция и ее регуляция в клетках млекопитающих». Тенденции биохимических наук . 32 (3): 111–17. дои : 10.1016/j.tibs.2007.01.003 . ПМИД 17291767 .
- ^ , Нориюки, Ютака; Мацуо, Томонори; Като, Киёси; Ито, Нозава, Ёсинори (февраль 2007 г.). Танака Митохондриальная гаплогруппа N9a защищена от метаболического синдрома» . Diabetes . 56 (2): 518–521. : 10.2337 /db06-1105 . PMID 17259400. . S2CID 34199769 doi
- ^ Теодорату, Европа; Дин, Фархат В.Н.; Фаррингтон, Сьюзен М.; Цетнарский, Розанна; Барнетсон, Ребекка А.; Портеус, Мэри Э.; Данлоп, Малькольм Г.; Кэмпбелл, Гарри; Тенеса, Альберт (февраль 2010 г.). «Связь между распространенными вариантами мтДНК и смертностью от колоректального рака или рака любой причины» . Канцерогенез . 31 (2): 296–301. дои : 10.1093/carcin/bgp237 . ПМИД 19945968 .
- ^ Гото, Ю. (сентябрь 1993 г.). «[MELAS (митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды): клинические особенности и мутации митохондриальной ДНК]». Нихон Ринсё. Японский журнал клинической медицины . 51 (9): 2373–8. ПМИД 8411715 .
- ^ Ахуджа, Абхиманью С. (21 мая 2018 г.). «Понимание митохондриальных миопатий: обзор» . ПерДж . 6 : е4790. дои : 10.7717/peerj.4790 . ПМЦ 5967365 . ПМИД 29844960 .
- ^ Ангиредди, Раджеш; Казми, Хасан Раза; Шринивасан, Сатиш; Сунь, Ли; Икбал, Джамиль; Фукс, Серж Ю.; Гуха, Манти; Кидзима, Такаши; Юэнь, Тони; Заиди, Моне; Авадхани, Нараян Г. (август 2019 г.). «Дисфункция цитохром-с-оксидазы усиливает фагоцитарную функцию и образование остеокластов в макрофагах» . Журнал ФАСЭБ . 33 (8): 9167–9181. дои : 10.1096/fj.201900010RR . ПМК 6662975 . ПМИД 31063702 .
- ^ Каррьери, Джузеппина; Бонафе, Массимилиано; Де Лука, Мария; Роза, Джузеппина; Варкасия, Октавия; Бруни, Амалия; Малетта, Рафаэле; Накмиас, Бенедетта; Сорби, Сандро; Корсонелло, Франческо; Ферако, Эмидио; Андреев Кирилл Ф.; Яшин Анатолий Игоревич; Франчески, Клаудио; Де Бенедиктис, Джованна (март 2001 г.). «Гаплогруппы митохондриальной ДНК и аллель APOE4 являются независимыми переменными при спорадической болезни Альцгеймера». Генетика человека . 108 (3): 194–198. дои : 10.1007/s004390100463 . ПМИД 11354629 . S2CID 6171041 .
- ^ Мартин-Хименес, Ребека; Люретт, Оливье; Эбер-Шатлен, Этьен (1 августа 2020 г.). «Повреждение митохондриальной ДНК, связанное с болезнью Паркинсона» . ДНК и клеточная биология . 39 (8): 1421–1430. дои : 10.1089/dna.2020.5398 . ПМИД 32397749 .
- ^ Чиннери, Патрик Ф; Эллиотт, Ханна Р.; Сайед, Анила; Ротвелл, Питер М. (май 2010 г.). «Гаплогруппы митохондриальной ДНК и риск транзиторной ишемической атаки и ишемического инсульта: исследование генетической ассоциации» . Ланцет Неврология . 9 (5): 498–503. дои : 10.1016/S1474-4422(10)70083-1 . ПМЦ 2855429 . ПМИД 20362514 .
- ^ Руис-Песини, Эдвард; Лапенья, Анна-Кристина; Тен-Санчес, Кармен; Перес-Мартос, Акцикл; Монтойя, Юлиус; Альварес, Генри; Диас, Майкл; Уррис, Энтони; Монторо, Луи; Лопес-Перес, Мануэль Х.; Энрикес, Хосе А. (сентябрь 2000 г.). «Гаплогруппы мтДНК человека, связанные с высокой или пониженной подвижностью сперматозоидов» . Американский журнал генетики человека . 67 (3): 682–696. дои : 10.1086/303040 . ПМЦ 1287528 . ПМИД 10936107 .
- ^ Куртенэ, Моник Д.; Гилберт, Джон Р.; Цзян, Лан; Каммингс, Анна С.; Галлинз, Пол Дж.; Кейвуд, Лаура; Рейнхарт-Мерсер, Лори; Фаззелл, Дениз; Кнебуш, Клэр; Ло, Рене; МакКоли, Джейкоб Л.; Джексон, Чарльз Э.; Перикак-Вэнс, Маргарет А.; Хейнс, Джонатан Л.; Скотт, Уильям К. (февраль 2012 г.). «Митохондриальная гаплогруппа X связана с успешным старением у амишей» . Генетика человека . 131 (2): 201–208. дои : 10.1007/s00439-011-1060-3 . ПМЦ 4834861 . ПМИД 21750925 .
- ^ Браун, WM (1 июня 1980 г.). «Полиморфизм в митохондриальной ДНК человека, выявленный с помощью эндонуклеазного анализа рестрикции» . Труды Национальной академии наук . 77 (6): 3605–3609. Бибкод : 1980PNAS...77.3605B . дои : 10.1073/pnas.77.6.3605 . ПМК 349666 . ПМИД 6251473 .
- ^ «Лаборатория Палео-ДНК – Судебно-медицинская служба» . Архивировано из оригинала 13 марта 2012 г. Проверено 13 июня 2012 г.
- ^ Стоун, Энн К.; Старрс, Джеймс Э.; Стоункинг, Марк (1 января 2001 г.). «Анализ митохондриальной ДНК предполагаемых останков Джесси Джеймса». Журнал судебной медицины . 46 (1): 173–6. дои : 10.1520/JFS14932J . ПМИД 11210907 . S2CID 6480921 .
- ^ Перейти обратно: а б Гилл, Питер; Иванов Павел Львович; Кимптон, Колин; Пирси, Ромель; Бенсон, Никола; Талли, Джиллиан; Эветт, Ян; Хагельберг, Эрика; Салливан, Кевин (февраль 1994 г.). «Идентификация останков семьи Романовых методом анализа ДНК». Природная генетика . 6 (2): 130–135. дои : 10.1038/ng0294-130 . ПМИД 8162066 . S2CID 33557869 .
- ^ Иванов Павел Львович; Уодхамс, Марк Дж.; Роби, Ронда К.; Холланд, Митчелл М.; Видн, Виктор В.; Парсонс, Томас Дж. (апрель 1996 г.). «Гетероплазмия последовательности митохондриальной ДНК у Великого князя Российского Георгия Романова устанавливает подлинность останков царя Николая II» . Природная генетика . 12 (4): 417–420. дои : 10.1038/ng0496-417 . ПМИД 8630496 . S2CID 287478 .
- ^ Эшдаун-Хилл, Джон (2013). Последние дни Ричарда III и судьба его ДНК . История Пресс. ISBN 978-0-7524-9205-6 . [ нужна страница ]
Дальнейшее чтение
[ редактировать ]- Ли, Сянци; Си, Цянь; Фан, Миншуан; Чжу, Чжао; Ван, Цзиннань; Чжан, Чжан; Ни, Минцзе; Гу, Минджун (2016). «Кратковременное лишение сыворотки не вызывает значительных мутаций митохондриальной ДНК в клетках гладких мышц сосудов, выявленных с помощью новой технологии секвенирования нового поколения . » Чаобао; Ху, Шуанган ; 48 (9): 862–4 doi : 10.1093/abbs/ gmw059 PMID 27261779 .
Внешние ссылки
[ редактировать ]- ISOGG YBrowse : браузер генома.
- mitoWheel : браузер митогенома.
- Митохондриальная ДНК в MedlinePlus
- MITOMAP , сообщает об опубликованных данных об вариациях митохондриальной ДНК человека.